
DNA, the blueprint of life, spans about 2 meters in length. This complexity is similar to stuffing an entire library into a shoebox without crumpling a single page. The complexity of this folding is a subject of immense scientific interest. Modeling this complexity is crucial for understanding gene expression, cellular function, and numerous other biological processes. The reflection of chromatin architecture is possible thanks to the recently developed MultiMM model by Sevastianos Korsak, Krzysztof Banecki, and Dariusz Plewczynski from the Warsaw University of Technology and the University of Warsaw. This paper explains the operations and importance of MultiMM in simulating chromatin.
What is Chromatin?
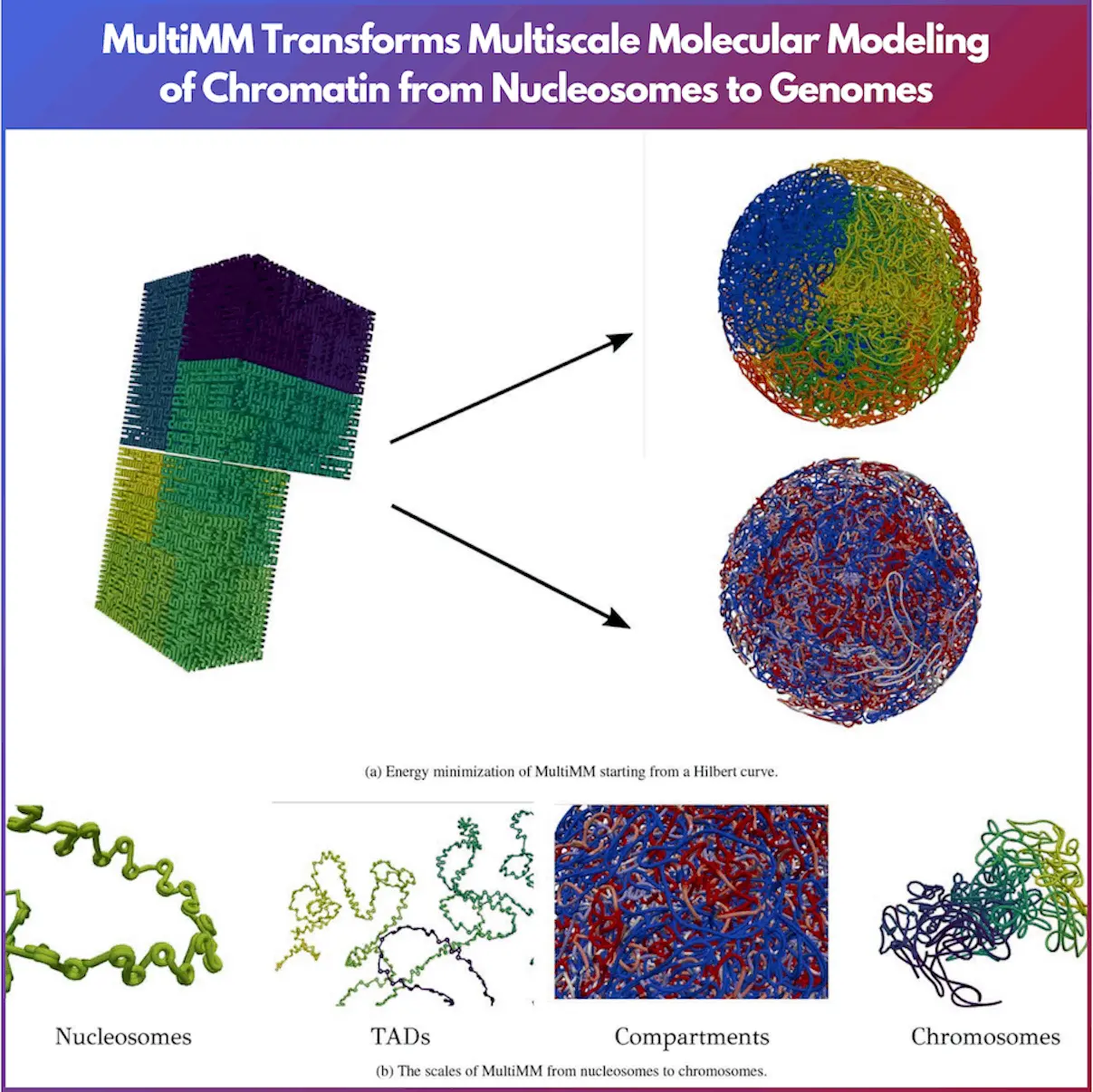
Chromatin’s fundamental units are nucleosomes, which are units of DNA wrapped around histone proteins. These nucleosomes form loops through interactions mediated by SMC (Structural maintenance of chromosomes) complexes and CTCF (a zinc-finger protein for transcription) proteins. These loops are further organized into compartments and sub-compartments, which assemble into entire chromosomes. The spatial arrangement and interactions within these structures play very important roles in regulating gene expression and other cellular functions.
Challenges in Chromatin Modeling
Studying the organization of chromatin implies constructing a hierarchical system, which is rather difficult in biophysics. Current approaches include some that work on a particular scale and some that require large computational power for the complexity of present-day systems. These drawbacks are solved by MultiMM for chromatin simulation.
Introduction to MultiMM
MultiMM is a tool built based on OpenMM, which aims to simulate the chromatin 3D structure, given the fact that chromatin is a multi-component and multi-length-scale structure. Starting from an initial configuration based on a Hilbert curve, MultiMM simulates the genome’s structure efficiently. Users can input files specifying nucleosome positioning, loops, compartments, or subcompartments, allowing for extensive customization.
How MultiMM Works
Input Parameters and Data
MultiMM requires minimal input data, including files containing loops, coordinates, and chromosomes for the region of interest. Users can adjust the simulation granularity and input compartment or subcompartment files to model block-copolymer attractions. Nucleosomes are depicted as static helical structures integrated into the final structure based on their density signals.
Implementation of the Model
The model begins by adapting the provided loop data to match the simulation’s granularity, consolidating loop strengths, and retaining only statistically significant loops. Compartmentalization data are imported and converted into a discrete value vector. The initial structure is constructed based on a Hilbert curve, providing a self-avoiding and compact starting point.
Energy Minimization
MultiMM uses a multiscale molecular force field, including strong harmonic bonds and angle forces between adjacent beads and harmonic spring forces, to model long-range loops. For compartment modeling, a block-copolymer potential with discrete energy levels is applied. The genome is folded within a spherical container, and lamina interactions are modeled with trigonometric potentials.
Output Files
After using MultiMM, output files with .cif extensions are created, which can be visualized using the pyvista library. Separate .cif structures for each chromosome are also extracted, allowing for detailed examination of individual chromosomal configurations.
The Advantages of MultiMM
User-Friendly: MultiMM is designed to be accessible to researchers with varying levels of expertise, prioritizing user experience.
Versatile: It accommodates a wide range of input data and simulation parameters, allowing for flexible and tailored simulations.
Efficient: By leveraging geometric modeling and energy minimization, MultiMM delivers fast and accurate simulations without requiring extensive computational resources. It runs the energy minimization with a graphic card or multiple CPU cores.
Future Prospects
As research into chromatin structure advances, tools like MultiMM will play an essential role in expanding our understanding. Its open-source nature and compatibility with various computational platforms ensure that it will remain a valuable resource in chromatin research.
Conclusion
MultiMM represents a considerable advancement in chromatin modeling. This is because it offers an efficient and versatile tool for simulating the intricate structure of chromatin. Its ability to model the genome with nucleosome-resolution structures provides researchers with a powerful means to explore and understand the complexities of chromatin folding and its implications for gene regulation and cellular function. By using MultiMM in bioinformatics, researchers can make many discoveries in genomics and molecular biology.
Article Source: Reference Paper | Open-source software and the manual are freely available on GitHub.
Important Note: bioRxiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Follow Us!
Learn More:




Neermita Bhattacharya is a consulting Scientific Content Writing Intern at CBIRT. She is pursuing B.Tech in computer science from IIT Jodhpur. She has a niche interest in the amalgamation of biological concepts and computer science and wishes to pursue higher studies in related fields. She has quite a bunch of hobbies- swimming, dancing ballet, playing the violin, guitar, ukulele, singing, drawing and painting, reading novels, playing indie videogames and writing short stories. She is excited to delve deeper into the fields of bioinformatics, genetics and computational biology and possibly help the world through research!











{kind=link}