
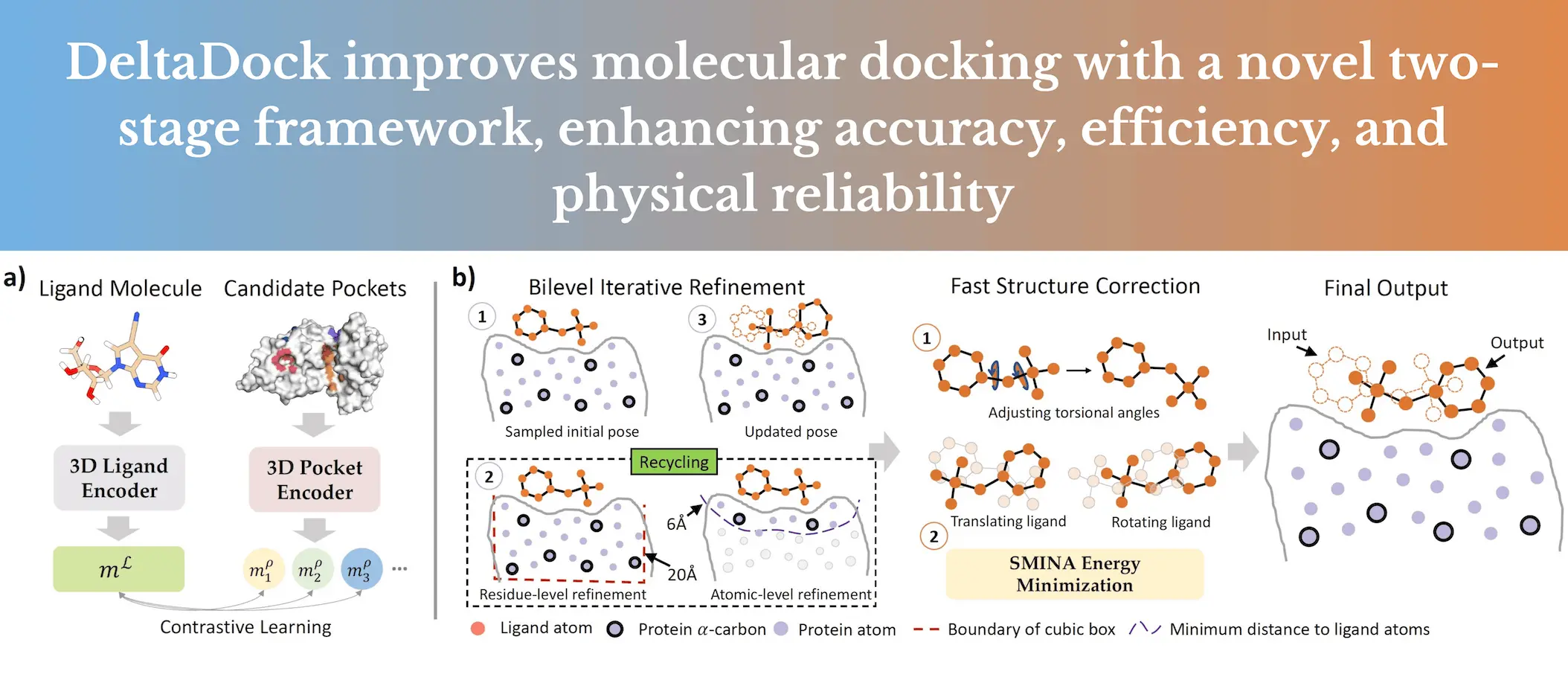
DeltaDock is a novel two-stage framework capable of performing molecular docking, which has not been achieved before. It does so by combining the strengths of geometric deep learning (GDL) methods with reliability associated with traditional sampling approaches. The DeltaDock framework incorporates in its architecture a pocket prediction stage followed by a site-centered docking stage. Researchers developed it at the State Key Laboratory of Cognitive Intelligence at the University of Science and Technology of China and the Center for Quantitative Biology at the Academy for Advanced Interdisciplinary Studies, Peking University.
Molecular docking involves the prediction of the interacting binding sites between small molecules, or ligands, and a macromolecule such as proteins, which is a very adage practice in discovering novel drug candidates and understanding the complexes formed in greater exquisite detail. Traditional docking methods rely on sampling techniques, which can be computationally expensive. Such methods require very high computer resources due to their reliance on sampling techniques. Recently, GDL, geometric deep learning, has developed a strong new potential that may be both quicker and more accurate.
A Two-Stage Approach for Molecular Docking
DeltaDock is a novel molecular docking framework consisting of two primary stages: pocket prediction and site-specific docking. In the first stage, DeltaDock utilizes a pocket-ligand contrastive alignment to determine the best possible binding site for the ligand of interest. This technique combines the use of ligand embedding and the embedding of candidate pockets to measure the similarity between them and choose the one with the optimal matching.
After a binding pocket has been determined, DeltaDock proceeds with stage two of the process of the docking: improvement of the binding pocket ligand pose. This refinement model is bi-level, encompassing both residue-level and atom-level refinement. DeltaDock uses constraints such as steric clashes, bond lengths and angles, and energy minimization to maintain the physical plausibility of predicted poses. This integral strategy allows DeltaDock to make specific and accurate binding pose predictions, adding value to the study of molecular docking and drug development.
How DeltaDock Works:
DeltaDock is a molecular docking tool, which works in two consecutive steps. It is made up of a pocket prediction stage and a site-specific docking stage.
Pocket Prediction Stage
- Candidate Pocket Generation: A set number of candidate pockets are produced using the existing pocket prediction methods such as DSDP, P2Rank, Fpocket, SiteMap, and DoGSite3.
- Ligand and Pocket Encoding: Ligand and each of the pockets are embedded through Attentive-FP (AFP) and Geometric Vector Perceptron, respectively (GVP).
- Contrastive Embedding Alignment: Cosine similarity measures the similarity between the ligand embedding and each pocket embedding. The pocket with the highest similarity is chosen as the predicted pocket.
The Site-Specific Docking Stage
- Initial Pose Sampling: An efficient GPU-accelerated algorithm places the initial pose for sampling.
- Bi-level Iterative Refinement: A bi-level E(3)-equivariant graph matching network (Bi-EGMN) will be used for the iterative smoothing of the pose. The bi-level strategy first delivers the refinement at the residue-level structure and then the atom-level structure. The bi-EGMN layer is responsible for coordinating the ligand coordinates per the protein-ligand interactions.
- Fast Structure Correction: The torsion alignment step eases the bridging of the plausibility between bond-flank lengths and bond angles. An energy minimization step using SMINA further refines the structure.
Overall Workflow:
1. Input: Protein and ligand structures.
2. Pocket prediction: Identify the most likely binding pocket.
3. Initial pose sampling: Generate an initial pose within the pocket.
4. Bi-level iterative refinement: Refine the pose iteratively.
5. Fast structure correction: Ensure the physical validity of the final pose.
6. Output: predicted binding pose.
By combining these components, DeltaDock provides a robust and accurate framework for molecular docking.
Experimental Results
DeltaDock was assessed using the PDBbind and PoseBusters datasets. It did better than baseline methods across both blind docking and docking at specific sites. In the case of blind docking, for instance, DeltaDock had a success ratio of 47.4%, which was an improvement of 31% over the previous sole holder of this domain, the GDL method, DiffDock.
Implications for Drug Discovery:
The use of DeltaDock offers a great deal of prospects in drug discovery. It has the potential to reduce the time it takes to identify viable drug candidates as it furnishes researchers with more accurate and quicker docking predictions. Besides this, it is capable of predicting physically valid poses for molecules, and therefore, most designs will turn out to be effective and with little side effects.
Conclusion
DeltaDock is one of the latest leaps forward in the science of molecular docking. The two-stage process, together with predicting accurate, efficient, and high-fidelity binding poses, adds considerable applicability value in drug design and many fields of biology.
As the investigation in this area develops further, we can expect to see even more powerful and sophisticated methods for understanding protein-ligand interactions.
Such developments are likely to include the following:
- Increasing the number of training data points for DeltaDock to enhance its performance further.
- Use DeltaDock simultaneously with other drug discovery processes, including virtual high-throughput screening and molecular dynamics modeling.
- Use DeltaDock approaches on other biological systems such as protein-protein interactions or enzyme-substrate complexes.
Overall, DeltaDock is a novel valuable tool with plausible applications in drug development and a significant contribution to biology.
Article Source: Reference Paper | All codes and data will be released on GitHub.
Important Note: arXiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Follow Us!
Learn More:




Anchal is a consulting scientific writing intern at CBIRT with a passion for bioinformatics and its miracles. She is pursuing an MTech in Bioinformatics from Delhi Technological University, Delhi. Through engaging prose, she invites readers to explore the captivating world of bioinformatics, showcasing its groundbreaking contributions to understanding the mysteries of life. Besides science, she enjoys reading and painting.











{kind=link}