
In synthetic biology, turnover numbers (kcat) — a critical measure of an enzyme’s efficiency—have a variety of applications. On the other hand, kcat measurement of enzymes takes time. Although prediction accuracy has increased thanks to deep learning models, more advancements are still required, particularly for enzymes whose sequences differ significantly from those in the training dataset. Here, scientists introduce DeepEnzyme, a state-of-the-art deep learning model that integrates the most recent Transformer and Graph Convolutional Network (GCN) models to extract protein sequence and three-dimensional structure information. DeepEnzyme is a unique approach that uses characteristics from both sequences and 3D structures to improve prediction accuracy and robustness in enzymes. DeepEnzyme’s performance is much enhanced by this method, especially when processing enzymes with little sequence similarity. It also makes it possible to assess how point mutations affect the catalytic activity of enzymes, determining which residue locations are essential for catalytic activity. This innovative discovery makes a substantial contribution to the comprehension of the roles played by enzymes and the patterns of evolution in different species.
Introduction
Under saturating conditions, the greatest number of substrate molecules that an enzyme may convert into a product in a unit of time is known as the enzyme turnover number (kcat).
These days, estimating kcat with accuracy has become essential for many applications, such as enzyme design and protein engineering. To build complex metabolic models that reflect the relationships between genotypes and phenotypes, measuring enzyme kcat values at the genome scale is also essential. Even with the abundance of kcat values readily available in databases, the overall number of enzymes with kcat values measured experimentally is still far fewer than the entire number of proteins that have been sequenced.
Understanding DeepEnzyme
The researchers introduce DeepEnzyme, a unique deep-learning algorithm that improves the accuracy of enzyme kcat predictions by utilizing protein sequences and three-dimensional structures. Transformers have shown remarkable effectiveness in protein language modeling, and DeepEnzyme combines them with Graph Convolutional Network (GCN), a tried-and-true method for extracting structural characteristics from proteins. With the help of this combination, DeepEnzyme can extract both structural and sequence properties from substrates and enzymes, giving it access to extensive biochemical data that can be used to learn about the catalytic function of enzyme proteins. DeepEnzyme shows better accuracy and robustness in kcat value prediction than current models. Consequently, DeepEnzyme can facilitate the opportunity for the logical optimization of enzyme activity in conjunction with cutting-edge molecular technologies and accelerate the functional investigation of hitherto unexplored enzymes.
Framework of DeepEnzymes
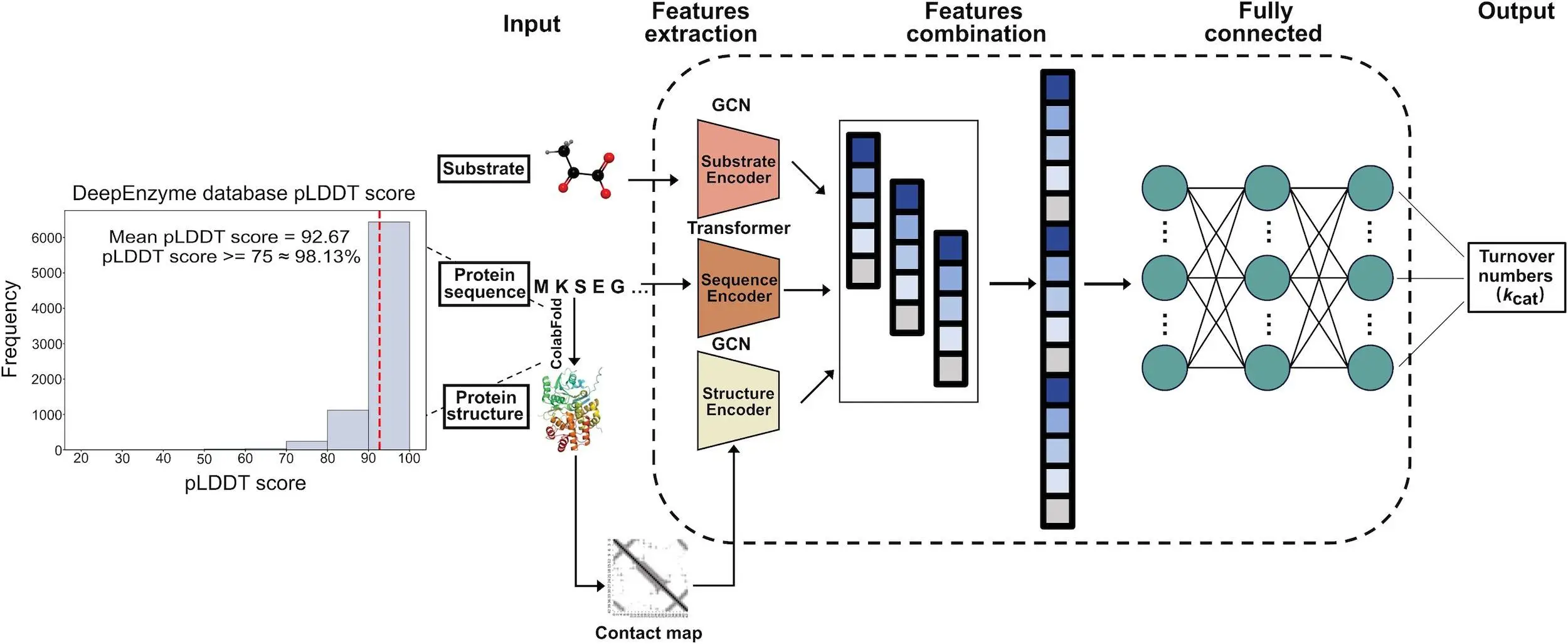
Utilizing characteristics from protein 1D sequence, 3D structure, and substrate, DeepEnzyme is a deep learning model for kcat prediction. Using the Cα − Cα < 10Å requirement, the 3D structure changes into a contact map, while the Transformer model pulls features from the protein 1D sequence. From protein 3D structures, GCN uses the contact map to derive structural information. Utilizing its SMILES formula, the RKit extracts substrate properties such as adjacency matrices and fingerprints. An extensive embedding vector is produced by the GCN module by combining feature vectors from the substrate and enzyme. With a neural attention technique based on representation vectors, the kcat value is anticipated. ColabFold is utilized to estimate the structures of enzymes without experimentally known 3D structures. All projected protein 3D structures have an average predicted local distance difference test score of 92.67, which guarantees high-quality structures for DeepEnzyme training.
DeepEnzymes shows improved efficiency of Kcat of 3D Structures
Protein sequence similarity between training, validation, and testing datasets has caused problems for DeepEnzyme, a deep learning model. A dataset free from overfitting and subpar generalisation was produced as a result of preprocessing the data to remove extremely similar sequences in order to solve this problem. In the dedicated training phase, DeepEnzyme achieved a high Pearson correlation coefficient (PCC) value of about 0.77 on the test dataset, indicating a relatively low prediction error. This preprocess allowed DeepEnzyme to train and assess well.
The prediction accuracy of DeepEnzyme, a machine learning model, was assessed by contrasting the values of predicted and measured kcat for enzymes belonging to different categories and sources. According to the study, wild-type enzymes only had a prediction accuracy (PCC) of 0.67, but mutant enzymes had a better PCC of 0.84. This implies that the amount of enzymes from each group may have an impact on the prediction’s accuracy. With better R2 values for enzymes from the EC 1 and EC 2 groups, DeepEnzyme also demonstrated significant prediction skills for enzymes of different EC numbers. The incorporation of structural data improved prediction accuracy significantly. DeepEnzyme computed the average R2 value on the testing dataset from five training rounds, obtaining a 0.58 R2 value to prevent bias in dataset sampling. In conclusion, DeepEnzyme does well in kcat prediction when protein 3D-structure features are taken into account; nevertheless, the bulk of the relevant data may have a detrimental impact on prediction accuracy.
DeepEnzyme: Performance Overview
- Despite sequence similarities, a deep learning model called DeepEnzyme has demonstrated a notable boost in prediction accuracy. The degree of similarity between the protein sequences in the test and training datasets has a major impact on the model’s performance. By preprocessing the sequence dataset, DeepEnzyme’s propensity to overfit during training is greatly reduced, enhancing the model’s capacity for generalization and guaranteeing accurate kcat predictions at various sequence similarity levels. DeepEnzyme performs better than earlier models, even with low sequence similarity (0–50%), highlighting the significance of identifying and using structural characteristics of the enzyme during training.
- DeepEnzyme may be useful in assessing the impact of saturation mutagenesis on the catalytic efficiency of enzymes. In this regard, two enzymes were used to illustrate DeepEnzyme’s functionality. The outcomes show that DeepEnzyme can reflect the ways in which single mutations impact the catalytic efficiency of an enzyme to a significant degree. Additionally, DeepEnzyme gives binding and active areas in a protein 3D-structure larger weight values. DeepEnzyme may facilitate the logical design of more effective enzymes because these functional areas are strongly connected to the activity and function of enzymes.
- Compared to previous models, the deep learning model DeepEnzyme has demonstrated better prediction accuracy in kcat analysis. The dataset employed, which had fewer than 12,000 enzyme-substrate pairings, may be improved, though. Important experimental characteristics like pH and temperature were not taken into account during the model’s training, which could have resulted in differences between the measured and projected kcat values. Furthermore, combining several sophisticated pre-training models into DeepEnzyme may improve prediction accuracy even more. In the future, protein engineering may proceed more quickly, thanks to this combination with other deep learning algorithms.
Conclusion
DeepEnzyme is a unique model that leverages deep learning architectures such as Transformer and GCN to improve enzyme kcat prediction. With far higher prediction accuracy, it extracts and aggregates key characteristics from the substrate, protein 1D sequences, and 3D structures. DeepEnzyme uses high-quality 3D structures to predict kcat better than earlier models. Outperforming TurNuP, DLKcat, and DLTKcat, it gets an impressively higher R2 value at ∼0.6 on the test dataset. DeepEnzyme does excellent in qualitatively predicting how single-point mutations impact enzyme catalytic efficiency.
Article Source: Reference Paper | The codes are accessible on GitHub
Follow Us!
Learn More:




Deotima is a consulting scientific content writing intern at CBIRT. Currently she's pursuing Master's in Bioinformatics at Maulana Abul Kalam Azad University of Technology. As an emerging scientific writer, she is eager to apply her expertise in making intricate scientific concepts comprehensible to individuals from diverse backgrounds. Deotima harbors a particular passion for Structural Bioinformatics and Molecular Dynamics.











{kind=link}