

Comprehending sequence–structure–function relationships is challenging for proteins with low similarity to existing proteins. New and improved alignment approaches are required for such proteins to find their similarity to previously annotated sequences. To close this gap, scientists from New York University, USA, developed two cutting-edge deep learning methods: TM-Vec and DeepBLAST. The search for structural similarities in massive sequence databases is made more accessible by TM-Vec, which precisely predicts TM-scores, a measure of structural similarity, directly from sequence data. On the other hand, DeepBLAST can align proteins solely based on sequence information by identifying structurally homologous regions. These techniques perform better than conventional sequence and structure-based alignment techniques. The effectiveness of TM-Vec and DeepBLAST on various datasets is discussed in this article, emphasizing how well they can recognize distantly related proteins, which is challenging for current state-of-the-art techniques.
Importance of Protein Sequences, Structures, and Functions
For Protein annotation, structure prediction, protein-protein interactions, protein design, and evolutionary study, understanding the functions and interactions of proteins is essential. Biotechnology heavily relies on proteins. Scientists have previously used sequence similarity to predict shared protein activities and infer protein homology. Proteins with a high degree of sequence similarity (>25%) can employ this tactic. The process of remote homology discovery, which involves identifying structurally related proteins with little sequence similarity, is difficult. It is challenging to identify structurally related proteins with little sequence similarity; this process is known as remote homology discovery. Current alignment methods based on sequence alignment are effective for closely related proteins, but alignment methods based on structural alignment are more effective for distantly related proteins.
By superimposing protein structures, structural alignment software like TM-align, Dali, FAST, and Mammoth calculates the structural similarity between two objects. However, there are drawbacks to these techniques, such as the fact that most proteins don’t have available structures and that using them at scale requires a lot of computer power.
A New Dawn with TM-Vec and DeepBLAST
TM-Vec was created to produce vector representations of proteins and produce precise structural similarity scores. It can produce indexable databases, allowing for effective protein-based structural similarity-based querying. TM-Vec in extensive structural similarity searches in databases like CATH and SWISS-MODEL, demonstrating its capacity to scale with database size while preserving high precision. On the other hand, DeepBLAST can calculate structural alignments between protein pairs based just on their sequences.
According to experiments on the Malidup and Malisam structure databases, it is capable of deriving structural alignments from distant homologs.
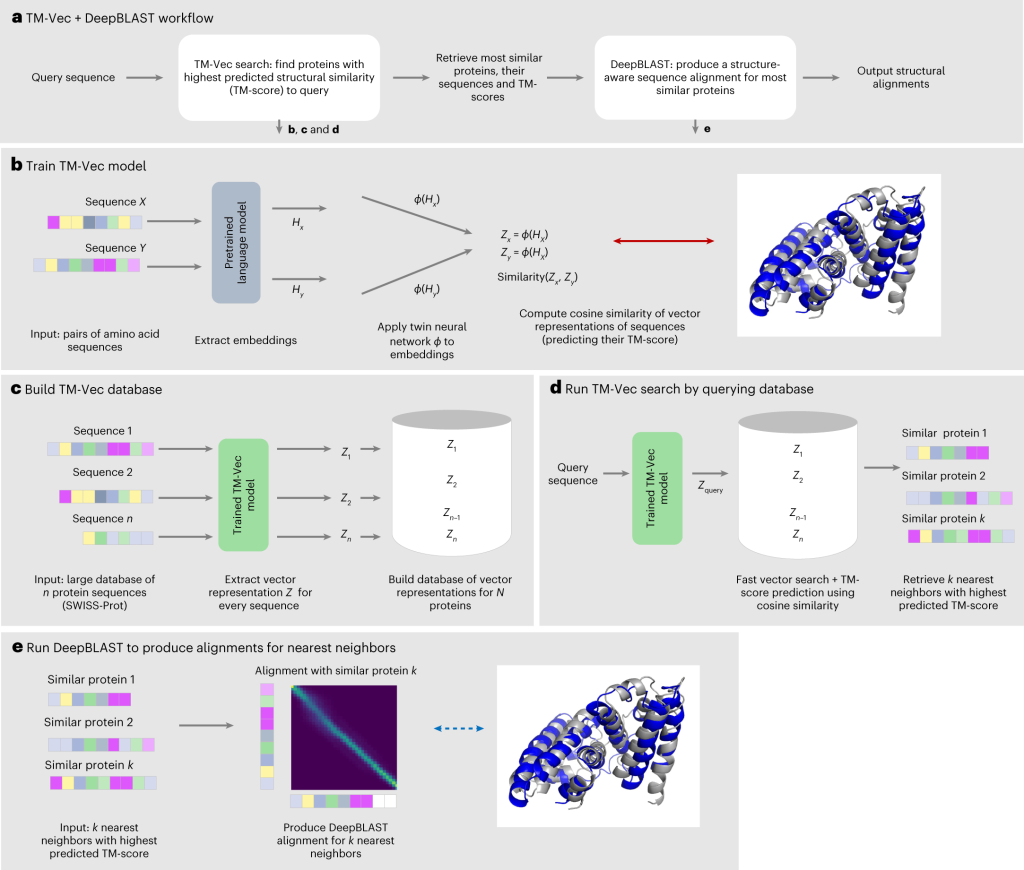
Image Source: https://doi.org/10.1038/s41587-023-01917-2
Rigorous Benchmarking on SWISS-MODEL and CATH Databases
To assess the viability of our TM-score prediction strategy, we subjected TM-Vec to rigorous benchmarking against the SWISS-MODEL and CATH databases. TM-Vec was trained using a significant amount of data in the SWISS-MODEL database, which consisted of over 150 million protein pairings (from 277,000 different SWISS-MODEL chains). Even for sequences with very little sequence identity, remarkably low prediction errors were seen—consistently.
In fact, TM-Vec outperformed conventional sequence alignment approaches in estimating structural differences between protein pairings with sequence identities < 0.1. The learned representations of TM-Vec outperformed those of other approaches that relied just on sequence or structure, demonstrating the method’s capacity to capture latent structural information. Researchers now have a strong tool for structural analysis across many biological tiers thanks to the visualized embeddings created by TM-Vec, which demonstrated its prowess in separating CATH structural classes.
It focused on three demanding scenarios—i) pairs that were never encountered together in training, ii) held-out domains, and iii) withheld folds—to further validate TM-Vec on the CATH protein domains. Strong performance was displayed by TM-Vec, which correctly predicted TM-scores for proteins. Notably, TM-Vec showed resilience to out-of-distribution findings, a critical skill for extrapolating beyond known protein structures, even while its prediction accuracy was reduced for proteins with TM-scores in the [0.75-1.0] range and held-out folds.
The learned representations created by TM-Vec were visualized and contrasted with alternative techniques relying solely on either sequence or structure. It showed that TM-Vec embeddings remarkably well represented hidden structural aspects of the CATH hierarchy. ProtTrans, a pre-trained language model, was outperformed by TM-Vec embeddings in the division of CATH structural classes into several tiers. Additionally, when the CATH database was encoded using TM-Vec, amazing results in the search were demonstrated, and TM-Vec accurately recovered proteins with the same fold in CATHS40 (88.1% accuracy) and CATHS100 (97% accuracy). The robustness and superior performance of TM-Vec in structural similarity prediction were demonstrated in head-to-head comparisons with other approaches, such as FoldSeek, MMseqs2, and ProtTucker, spanning several levels of the CATH hierarchy.
TM-Vec’s companion- DeepBLAST
DeepBLAST is excellent at obtaining structural alignments from protein sequences in addition to predicting structural similarity. In order to demonstrate DeepBLAST’s better performance in sequence-based alignment, it was compared to a number of sequence alignment techniques and four structural alignment techniques. Notably, DeepBLAST is a game-changer in the field of structural biology since it can find structural alignments for protein pairings with 25% sequence identity, which is a known issue for sequence alignment techniques.
The researchers used search real-time benchmarks to show the scalability of TM-Vec and found that it was effective at querying huge databases and encoding protein sequences. A useful tool for large-scale protein database searches, TM-Vec’s search runtimes surpassed BLAST and, significantly, TM-Vec obtained a ten times speedup compared to BLAST for specified circumstances.
The structural alignment runtime benchmarks of DeepBLAST showed its promise for large-scale structural alignment problems by highlighting its capacity to execute numerous alignments concurrently on a single GPU. To demonstrate TM-Vec’s competitive outcomes when exploring contemporary protein collections, the researchers assessed its performance on the DIAMOND benchmark.
Charting the Course for Future Protein Analysis
In conclusion, the development of TM-Vec and DeepBLAST represents a revolutionary advance in TM-score-based protein structure prediction and search. The usefulness of TM-Vec in identifying structural similarities, even among proteins with previously unidentified structures and folds, has been established through thorough benchmarking and validation on various datasets, including SWISS-MODEL, CATH, and MIP. The extraordinary precision and sensitivity of these techniques allow researchers to study the structural interactions between proteins with unmatched accuracy. TM-Vec and DeepBLAST have great potential for advancing structural biology and biotechnology research, ultimately advancing the comprehension of the complex world of proteins because of their scalability and adaptability.
TM-Vec is available on GitHub at https://github.com/tymor22/tm-vec, and DeepBLAST is available on GitHub at https://github.com/flatironinstitute/deepblast
Article Source: Reference Article
Learn More:




Prachi is an enthusiastic M.Tech Biotechnology student with a strong passion for merging technology and biology. This journey has propelled her into the captivating realm of Bioinformatics. She aspires to integrate her engineering prowess with a profound interest in biotechnology, aiming to connect academic and real-world knowledge in the field of Bioinformatics.





{kind=link}