
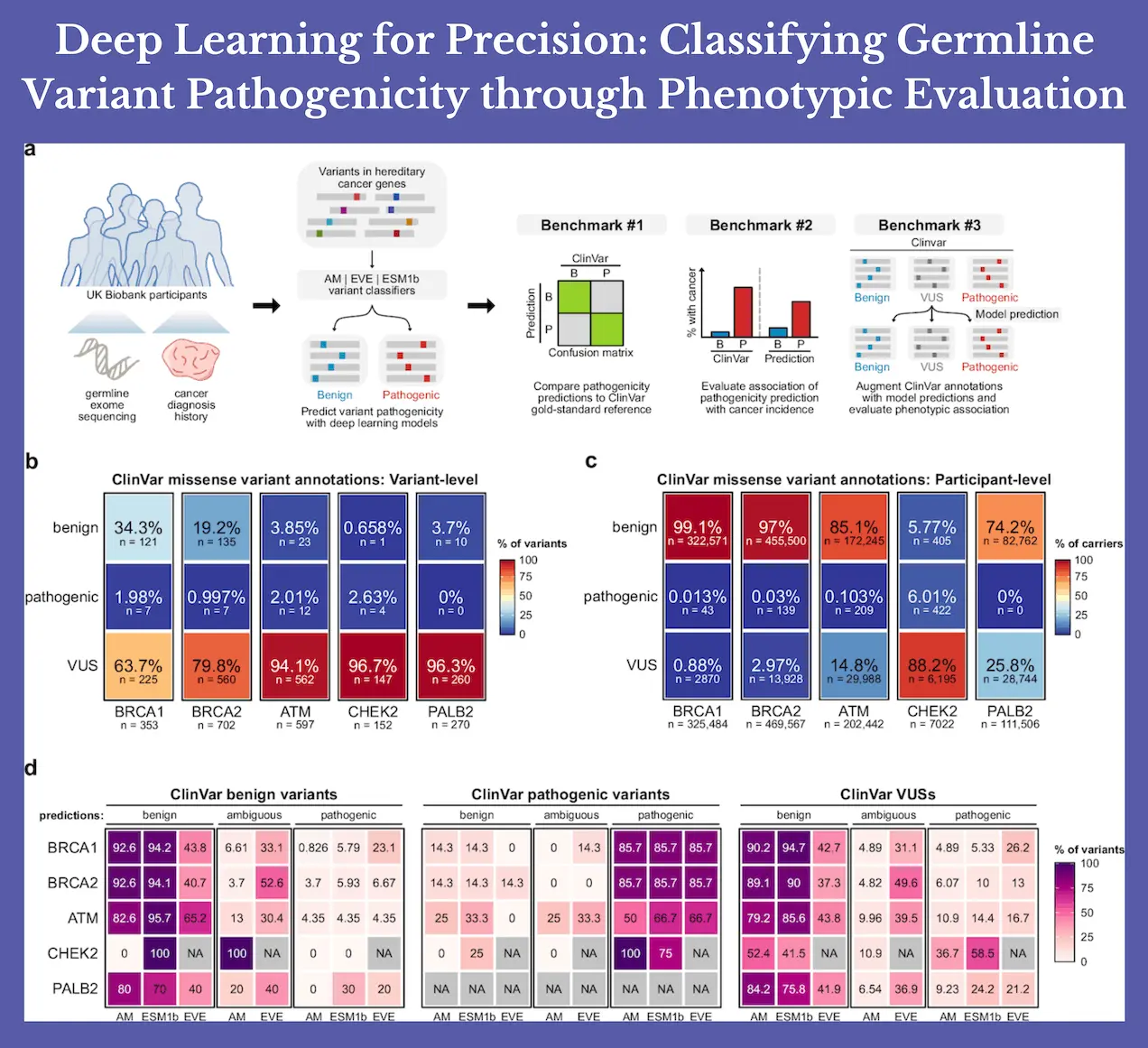
Deep learning models have been used to predict the pathogenicity of mutations in hereditary breast cancer gene variants in the UK Biobank. Here, researchers classified germline missense mutations in UK Biobank individuals using three state-of-the-art deep learning models for pathogenicity prediction. The findings demonstrated that missense mutations in BRCA1, BRCA2, and PALB2 were linked to an increased risk of breast cancer; however, when applied to variants of unknown significance, ATM and CHEK2 had little therapeutic value.
Introduction
Determining which variations are harmful and which are benign is a fundamental problem in germline genetic testing. Many changes are considered variations of unknown significance (VUSs) because they lack sufficient characterization. Annotations of variation pathogenicity from clinical references such as the ClinVar database can be independently recapitulated using deep learning models. The pathogenicity of missense variations that are not yet identified can also be predicted by these models, which could improve the present variant classification strategy. However, there hasn’t been a thorough assessment of these models concerning clinical illness characteristics. Therefore, determining whether deep learning models can meaningfully discriminate variations with higher risk of disease is important for clinical application, as clinical guidelines discourage the use of computational pathogenicity predictions alone.
3 State-of-the-art Deep Learning Models
Here, germline missense mutations in people in the UK Biobank were categorized using three state-of-the-art deep learning models for pathogenicity prediction (AlphaMissense, EVE, and ESM1b). For each of the five hereditary breast cancer genes (BRCA1, BRCA2, ATM, CHEK2, and PALB2), researchers benchmarked model-based pathogenicity predictions to see if existing deep learning models could accurately classify pathogenic germline variants that are functionally linked to an increased risk of breast cancer in the real world. Researchers additionally assessed pathogenicity predictions for BRCA1 and BRCA2 in connection to ovarian/fallopian malignancies to extend the research to another cancer with well-established hereditary risk genes.
Findings of the Study
Researchers show that, in a real-world setting, model-based pathogenicity predictions were linked to human illness characteristics, highlighting their potential for therapeutic use. Pathogenicity prediction models, in particular, have the potential to close important knowledge gaps by providing guidance on variant classification for genes with less research.
While none of the detected PALB2 variants were classified as pathogenic ClinVar, AlphaMissense, and ESM1b were able to identify variants that were associated with an elevated risk of breast cancer. This provides a tangible illustration of how deep learning models could influence variant classification within the framework of ACMG/AMP guidelines, thereby influencing clinical decision-making.
Furthermore, the fraction of those categorized as VUS carriers decreased with the use of composite classifiers. In situations where decreasing the percentage of VUS carriers is the top objective, such a compromise may be justified, even though this decrease in indeterminant categories typically comes at the expense of discriminative power.
The findings also show that there is still a significant barrier to widespread clinical implementation due to differences in model performance among genes. Researchers show that a scanning strategy to adjust gene-specific score thresholds could improve the specificity of AlphaMissense for detecting functional pathogenic mutations, thereby addressing the problem of gene-wise variation in model performance. This variance in the performance of the model could be partially attributed to underlying biases in the training data, which could lead to a relative underrepresentation of particular protein archetypes and harmful variations.
Furthermore, a significant percentage of missense variants in genes may confer vastly differing degrees of related cancer risk, and functionally detrimental mutations may also have minimal disease penetration, making it difficult to classify pathogenicity. The aforementioned phenomena have a strong known history with CHEK2, and they could potentially shed light on the conclusions on the restricted effectiveness of deep learning models in classifying CHEK2 missense variants.
Advantages of the Current Model Over Prior Studies
Previous research has frequently evaluated the model’s effectiveness by comparing its ability to reproduce ClinVar’s benign and pathogenic categories. This method’s design makes it impossible to critically assess model predictions for VUSs, which is arguably the main reason why deep learning models were created. Rather than focusing on a specific disease characteristic, such as breast cancer diagnosis, the work directly benchmarked model performance.
This allowed for a focused evaluation of whether the models could accurately classify VUSs. In light of this, researchers discovered that current deep learning models would not be useful for variable categorization in the context of ACMG/AMP guidelines because they have low clinical utility for predicting VUS pathogenicity in the majority of the hereditary cancer genes examined here. With the exception of PALB2, VUSs that were projected to be pathogenic often did not raise the risk of cancer in comparison to predicted benign VUSs.
Conclusion
This study emphasizes the need for clinicogenomic data in assessing the clinical usefulness of pathogenicity prediction algorithms in real-world settings. There is a drawback to using ClinVar and related variant databases as the gold standard reference for model evaluation: they have the potential to reinforce racial and ethnic differences already present in the literature. For example, compared to people of European heritage, those of African and Asian descent are more likely to have VUSs. As a result, when applied to other populations, pathogenicity prediction algorithms trained on current variation databases may perform less well. Going forward, researchers believe that the next generation of pathogenicity prediction models will be much enhanced by the early addition of more heterogeneous clinicogenomic populations.
Article Source: Reference Paper | Custom analysis code has been deposited to Github.
Follow Us!
Learn More:




Deotima is a consulting scientific content writing intern at CBIRT. Currently she's pursuing Master's in Bioinformatics at Maulana Abul Kalam Azad University of Technology. As an emerging scientific writer, she is eager to apply her expertise in making intricate scientific concepts comprehensible to individuals from diverse backgrounds. Deotima harbors a particular passion for Structural Bioinformatics and Molecular Dynamics.











{kind=link}