
The Central Dogma of Biology describes the process of DNA (Deoxyribonucleic acid) undergoing transcription to form RNA (Ribonucleic acid) and transformation to form proteins finally. However, the interactions between proteins and RNA are also crucial in the optimal operation of various processes in organisms. Improving computational prediction of RNA-protein interactions (RPIs) is important for advancing understanding and supporting experimental research across diverse species. Researchers from Friedrich Schiller University, the European Virus Bioinformatics Center, and the German Centre for Integrative Biodiversity Research, Germany, highlight tools that can predict RNA-protein interactions (RPIs) without requiring high-throughput data as input. They also provide an overview of prediction tools for RPIs, comparing them based on input, usability, and output.
What are RNA-Protein Interactions?
RPIs are fundamental to many molecular processes within biological entities, such as gene expression, RNA processing, modification, and degradation. RNA-binding proteins (RBPs) play a pivotal role in these interactions. The importance of RPIs is noted by their involvement in regulating protein synthesis, especially through interactions with messenger RNAs (mRNAs), microRNAs (miRNAs), and long non-coding RNAs (lncRNAs). Due to the extensive variety of processes influenced by RPIs, their dysfunctions are often linked to various diseases.
Despite the substantial understanding of RBPs in eukaryotic systems, especially within humans, there remains a sparsity of data for bacteria and viruses. For instance, while around 1,500 RBPs are annotated in humans, only approximately 180 are known for a typical bacterium, and intra-viral RPIs are less explored than host-virus interactions. RPIs often involve dynamic conformational changes and specific or non-specific binding of RNA molecules by proteins facilitated by various residues and structural motifs.
How are RPIs detected experimentally?
In experimental detection of RPIS, researchers can focus on an RNA molecule of interest to characterize potential proteins binding to it. These methods have their own challenges, though. Techniques like RNA affinity purification and RNA immunoprecipitation (RIP) combined with high-throughput sequencing (RIP-Seq) or crosslinking immunoprecipitation (CLIP) have advanced our understanding but have limitations like noise issues, co-immunoprecipitation of unwanted RBPs, and potential biases due to experimental conditions. For example, HITS-CLIP enables large-scale RPI detection but may lead to genetic mutations because of UV radiation utilized in crosslinking. Despite those challenges, in-vivo strategies typically offer greater biologically precise interaction facts. However, these methods may be costly and time-ingesting. Hence, experimental approaches can be supplemented with bioinformatics methods, which are discussed below.
Computational Prediction of RPIs
Algorithms use experimental data to analyze sequences and structures, predict binding sites, and classify RNA-protein interactions. Input for the algorithms is mostly the sequence or PDB (Protein Data Bank) 3D structure of either or both RPI partners. Various tools exist with different features like background data, machine-learning algorithms, and output types, utilizing experimental data for predicting RNA-protein interactions. Most of the time, though, this input data is unavailable. Hence, these tools can be broadly categorized based on their input requirements and the level of detail in their output. Some tools require high-throughput sequencing (HTS) data, while others do not, making them suitable for studies involving non-model organisms or specific RNA-protein complexes. HTS data is the result of analysis of dozens, hundreds, or even thousands of samples per day in a given laboratory or on a particular instrument. The researchers provide a comparative study of RPI prediction tools that do not need experimental HTS data as input.
Evaluation and Results
Researchers tested 30 algorithms on four examples from different organisms to see if they favor specific groups, providing insights into how well these algorithms predict RNA-protein interactions. They examined
(i) the human protein LARP7 binding to the 7SK snRNA;
(ii) the MS2 phage coat protein interacting with an RNA hairpin in the phage’s 165 genomes;
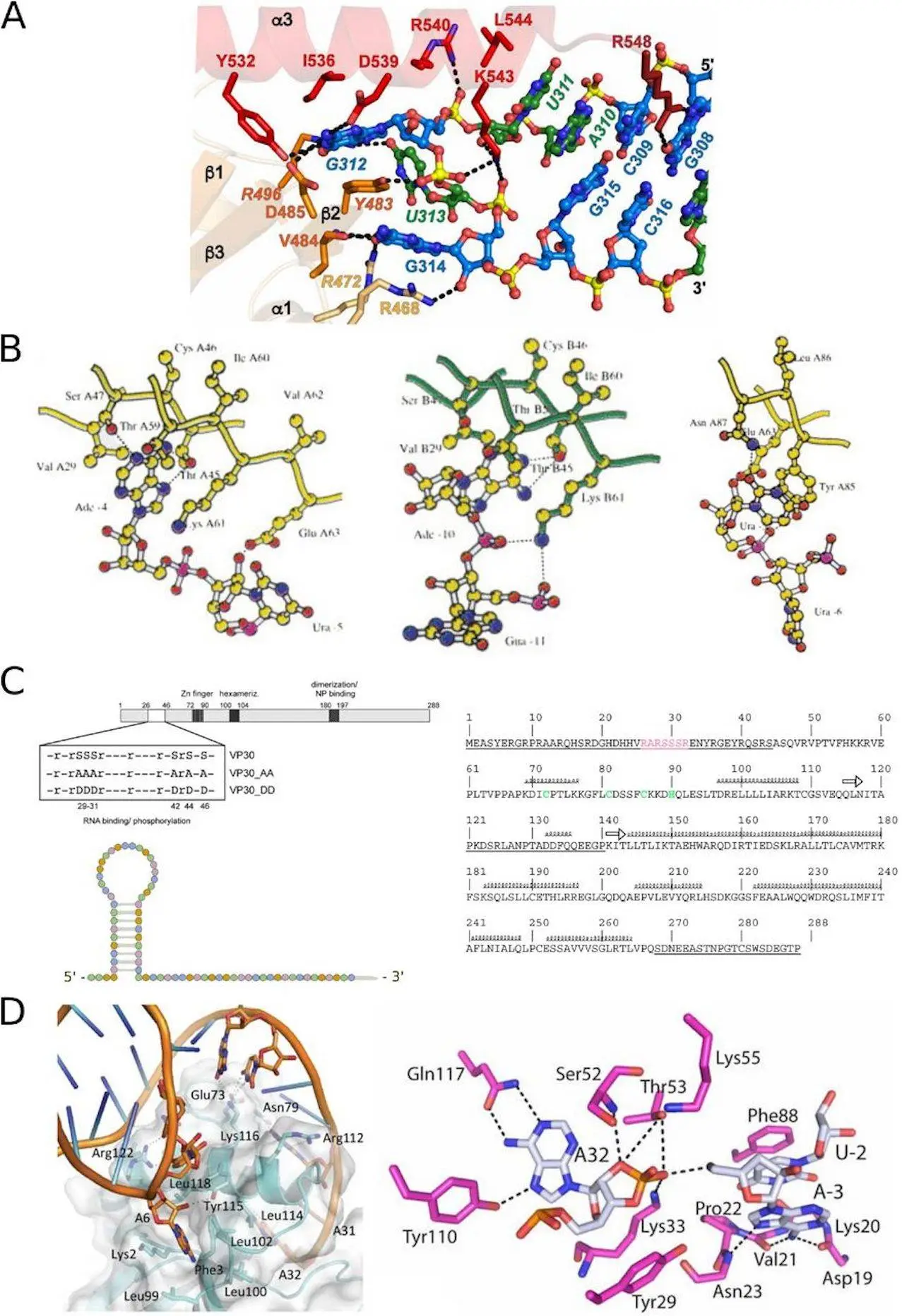
(iii) the Ebola virus VP30 protein binding to the viral RNA leader region; and
(iv) the bacterial toxin-antitoxin system ToxIN.
The ground truths were available for these known RPIs.
De novo RPI prediction tools need sequence or structure input of the potentially interacting RNA or protein molecules. The researchers have compiled an extensive overview of available RPI prediction tools at GitHub to aid users in RPI prediction analysis. Here are some of the tools:
RNA Sequence Input Tools: MEME, GraphProt, iDeepS.
Protein Sequence Input Tools: hybridNAP, DRNAPred, aaRNA, aPRBind, GraphBind, NucleicNet, KYG, PLIP.
RNA and Protein Sequence Input Tools: XRPI, RPISeq, IPMiner, PRIdictor, catRAPID.
RNA and Protein Structure Input Tools: PRIME3D2D, RSiteDB, DeepSite, PredPRBA, 3dRPC, COACH, OPRA, PRIHotscore.
The results show that the Ebola virus VP30 protein binds to RNA, regulating virus transcription. Tools like COACH and OPRA perform well in predicting interactions in the Ebola virus example. The bacterial ToxIN system involves RPIs forming a complex to neutralize toxin proteins—tools like PLIP and PRIHotscore excel in predicting RPIs in the ToxIN system.
Conclusion
The comparative analysis by the researchers shows us the importance of computational tools as a substitute for experimental approaches in RPI prediction, especially when high-throughput data is unavailable. These tools enhance our understanding of RPIs and facilitate targeted experimental research by providing preliminary interaction data.
Article Source: Reference Paper | Code is available on GitHub.
Important Note: bioRxiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Follow Us!
Learn More:




Neermita Bhattacharya is a consulting Scientific Content Writing Intern at CBIRT. She is pursuing B.Tech in computer science from IIT Jodhpur. She has a niche interest in the amalgamation of biological concepts and computer science and wishes to pursue higher studies in related fields. She has quite a bunch of hobbies- swimming, dancing ballet, playing the violin, guitar, ukulele, singing, drawing and painting, reading novels, playing indie videogames and writing short stories. She is excited to delve deeper into the fields of bioinformatics, genetics and computational biology and possibly help the world through research!











{kind=link}