
CancerVar leverages machine learning frameworks to evaluate the potential relevance of somatic cancer mutations in terms of cancer diagnosis, prognosis, and targetability, in addition to simply discovering them.
Researchers at Children’s Hospital of Philadelphia (CHOP) developed CancerVar, a novel machine learning empowered tool to interpret somatic variants in cancer. CancerVar can be accessed either as a web server or a command-line software tool, giving polished and semiautomated clinical interpretations of somatic cancer variants.
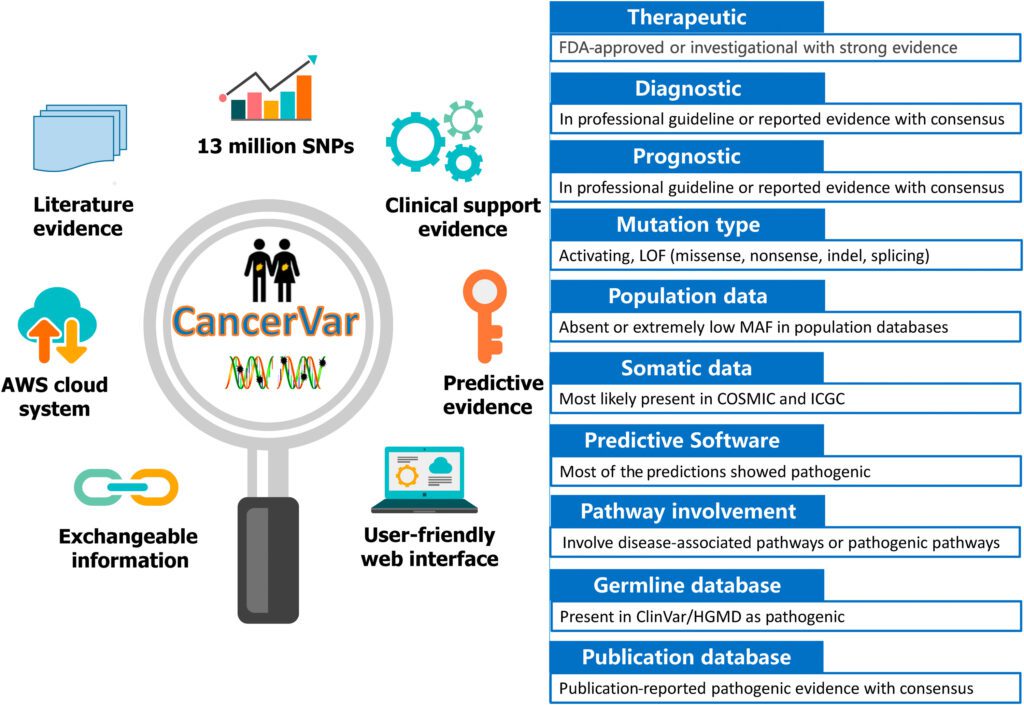
Image Source: CancerVar: An artificial intelligence–empowered platform for clinical interpretation of somatic mutations in cancer
Numerous knowledge bases are curated manually for supporting clinical interpretations of thousands of areas of interest in somatic mutations of cancer.
CancerVar will not replace human interpretation in a clinical setting, but it will significantly reduce the manual work of human reviewers in classifying variants identified through sequencing and drafting clinical reports in the practice of precision oncology. CancerVar documents and harmonizes various types of clinical evidence including drug information, publications, and pathways for somatic mutations in detail. By providing standardized, reproducible, and precise output for interpreting somatic variants, CancerVar can help researchers and clinicians prioritize mutations of concern.
Kai Wang, PhD, Professor of Pathology and Laboratory Medicine at CHOP
In any case, discrepancies or, in any event, conflicts of interpretations are seen among the knowledge bases. Besides, numerous undocumented mutations might clinically or functionally affect cancer but are not systematically interpreted by existing knowledge bases.
Somatic variant classification and interpretation are the most time-consuming steps of tumor genomic profiling. CancerVar provides a powerful tool that automates these two critical steps. Clinical implementation of this tool will significantly improve test turnaround time and performance consistency, making the tests more impactful and affordable to all pediatric cancer patients.
Marilyn M. Li, MD, Professor of Pathology and Laboratory Medicine, Director of Cancer Genomic Diagnostics
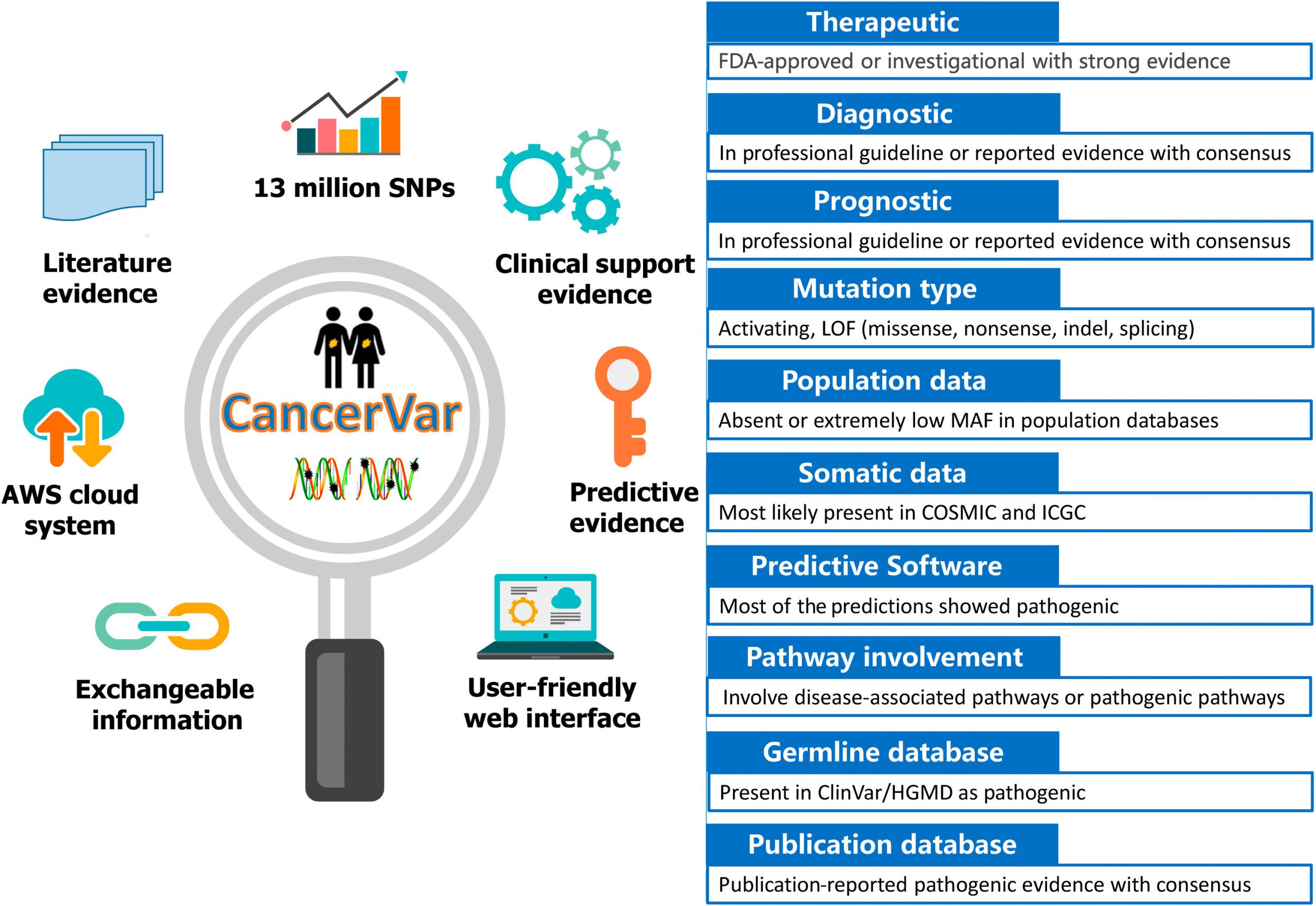
To address these difficulties, the scientists developed CancerVar to facilitate automated and standardized interpretations for 13 million somatic mutations given the AMP/ASCO/CAP 2017 guidelines.
The scientists further introduced a deep learning framework for the prediction of oncogenicity for the variants by the utilization of both functional and clinical features.
CancerVar accomplished satisfactory execution when contrasted with a few autonomous knowledge bases and, utilizing clinically curated datasets, demonstrated practical utility in the classification of somatic variants.
In outline, by the integration of clinical guidelines, with a deep learning framework, CancerVar facilitates clinical interpretations of somatic variants, lessens manual work, further develops consistency in variant classification, and advances implementation of the guidelines.
The AMP/ASCO/CAP 2017 Guidelines
Next-generation sequencing (NGS) has recognized countless somatic variants during clinical oncology practice to aid precision medicine.
To more readily comprehend the clinical effects of somatic mutations in cancer, a few knowledge bases have been arranged, including OncoKB, My Cancer Genome, CIViC, Precision Medicine Knowledge Base, JAX Clinical Knowledgebase (CKB), and Cancer Genome Interpreter.
Albeit clinically relevant, understanding somatic variants is not a standardized practice, and different clinical groups frequently produce unique or, in any event, conflicting outcomes.
To standardize the clinical interpretation of somatic variants in cancer and support effective clinical decision making, the Association for Molecular Pathology (AMP), American Society of Clinical Oncology (ASCO), and College of American Pathologists (CAP), together with proposed standards and guidelines for the understanding and detailing of somatic variants, ordering substantial variations into four levels:
- Strong clinical significance (level I),
- Potential clinical significance (level II),
- Uncertain significance (level III), and
- Benign (level IV).
These AMP/ASCO/CAP 2017 rules integrate 12 bits of evidence, including:
- Diagnostic, Prognostic, and Therapeutic clinical significance;
- Mutation type;
- Variant allele fraction [mosaic variant frequency (potentially somatic) and non-mosaic variant frequency (potentially germline)];
- Population information bases;
- Germline databases;
- Somatic databases;
- Predictive results of various computational algorithms;
- Pathway involvement; and
- Publications.
Discordance in Agreement Rates of Reclassification of Variants
Notwithstanding, as the AMP/ASCO/CAP classification scheme heavily depends on published clinical evidence for a given variant, ambiguous assignments among human curators regularly happen while involving similar evidence for a given variant.
The first overall observed agreement was just 58%; while giving similar evidential information of variants to the pathologists, the agreement rate of reclassification expanded to 70%.
The reasons behind such discordance are as per the following:
- Gathering data/evidence is complicated and may not be reproducible by a similar interpreter at various time slots;
- Various scientists might utilize different algorithms, cutoffs, and parameters, rendering interpretation less reproducible; and
- Recently published evidence for certain variants may not be integrated into the evaluation system immediately and systematically, which is particularly relevant for variants of unknown significance (VUSs).
Standardization of Interpretation of Somatic Variants
For the standardization of the interpretation of somatic variants across multiple knowledge bases, an all the more recently published knowledge base, MetaKB from The Variant Interpretation for Cancer Consortium, has accumulated evidence in view of the AMP/ASCO/CAP 2017 guidelines.
In any case, this MetaKB knowledge base likewise has the accompanying constraints:
- It just focuses on consensus interpretations for a limited number of known hotspot mutations, to such an extent that an enormous number of variants are presently classified as unknown clinical significance, however, they might be oncogenic through “loss of function” or “gain of function” in cancer;
- It just gives a summed up classification to every variant without demonstrating itemized evidence exhaustively while mapping the 12 criteria of the AMP/ASCO/CAP 2017 guidelines, and along these lines, users can’t conduct customized assessments in view of their protocols, experiences, and updated clinical knowledge; and
- It utilizes a straightforward scoring system to rank driver mutations disregarding heterogeneity of functional consequences of variants, particularly for recently identified variants detailed in the literature.
A Requirement for a Reliable and Accurate Computational Technique for Clinical Interpretations of Somatic Variants in Cancer
In clinical practice, when a somatic mutation is thought to have strong confidence in causing a functional effect on protein changes, clinicians probably interpret it with clinical significance or likely clinical significance. considered
Albeit various useful software tools, particularly sorting intolerant from tolerant (SIFT), PolyPhen-2, and functional analysis through hidden Markov models (FATHMM), have been created to predict functional effects, conflicts concerning specific mutations are consistently observed.
A portion of these devices utilize similar background data in light of alignment, evolutionary conservation, and homology, like MutationAssessor, FATHMM, cancer-specific high-throughput annotation of somatic mutations (CHASM) in view of random forest (RF) classifier, and CanDrA in light of support vector machine (SVM); others use consensus data by coordinating numerous sources of data from numerous computational tools, for example, combined tool adjusted total CTAT-cancer.
Albeit some meta-examination tools, for example, deleterious annotation of genetic variants using neural networks (DANN) and DriverPower, were subsequently evolved to focus on functionally significant variants utilizing more comprehensive functional scoring features as input, they have limitations in jointly modeling clinical impact features in view of the AMP/ASCO/CAP guidelines.
Since the guidelines will quite often be conservative (“negative diagnosis” is preferred to “wrong diagnosis”), a greater number of variants than predicted were misinterpreted as VUSs.
Moreover, the AMP/ASCO/CAP guidelines just designate seven functional impact prediction tools, like MutationAssessor, as the officially suggested tools, and just the variant from majority voting can be thought of “with clinical significance,” which misrepresents the heterogeneity of predictions of functional consequence in cancer progression.
Although utilizing existing tools might be valuable in the prediction of the overall impacts of cancer driver genes, it may not be ideal for focusing on novel mutations in these genes.
Notwithstanding the in silico predictive techniques above, interactome network approaches stand out for identifying oncogenic variants in cancer through genotype-phenotype studies.
In interactome networks, certain perturbed mutations (network hubs) can upset specific signaling pathways and protein-protein interactions (PPIs), bringing about similar cancer phenotypes in various patients.
These perturbed mutations, named “edgetic” mutations, are functionally significant but are understudied with existing cancer variant interpretation tools.
To address these difficulties and improve automated clinical interpretations of somatic variants in cancer, there is a major need for strong and accurate computational strategies utilizing both clinical evidence and functional impact score features.
The CancerVar Web Server
The scientists recently developed the standalone software named VIC written in Java, which was among the primary tools for interpreting clinical effects of somatic variants utilizing a rule-based scoring system based on 12 criteria of the AMP/ASCO/CAP 2017 guidelines.
In the ongoing study, the researchers developed an improved somatic variant interpretation tool called CancerVar (cancer variant interpretation) implemented in Python with a web server.
Contrasted with VIC, CancerVar is a particularly improved tool giving more choices to users:
- Python execution gives greater flexibility to integrate CancerVar into custom command-line work processes;
- CancerVar includes an easy to understand web server with precomputed clinical evidence for 13 million variants from 1911 cancer census genes through literature mining and database aggregations;
- They utilized a flexible AMP/ASCO/CAP rule-based score framework and a deep learning-based scoring framework that considers further developed interpretations; and
- RESTful application programming interface (API) is utilized to empower developers to access compiled knowledge.
CancerVar permits users to query clinical interpretations for variants utilizing the chromosome position, cDNA change, or protein change and intuitively adjust loads of scoring features in view of prior knowledge or additional user-specified criteria.
The CancerVar web server generates automated text with summed up illustrative interpretations, like diagnostic, prognostic, targeted drug responses, and clinical trial information for numerous hotspot mutations, altogether lessening the workload of human reviewers in drafting clinical reports in the practice of precision oncology.
The Endpoint
Clinical interpretation of cancer somatic variants is significant for clinicians and researchers working in the field of precision oncology, particularly given the transition from panel sequencing to whole exome/genome sequencing in cancer genomic studies.
This tool shows how we can use computational tools to automate human generated guidelines, and also how machine learning can guide decision making. Future research should explore applying this framework to other areas of pathology as well.
Kai Wang, PhD, Professor of Pathology and Laboratory Medicine at CHOP
To assemble a standardized, quick, and easy-to-use interpretation tool, the scientists developed command-line software tools along with a web server to evaluate the clinical effects of somatic variants utilizing the AMP/ASCO/CAP 2017 guidelines.
CancerVar is an upgraded version of the cancer variant knowledge base incorporated from previously developed tools for variant annotations and prioritizations, including ANNOVAR, InterVar, VIC, and iCAGES, as well as assembling existing variant annotation knowledge bases, for example, CIViC, CKB, and OncoKB.
In conclusion, CancerVar is both a web server and a command-line software tool that gives polished and semiautomated clinical interpretations for somatic variants in cancer.
In addition, CancerVar works with drafting clinical reports semi-automatically for panel sequencing, exome sequencing, or genome sequencing in cancer.
Article Source: Quan, L., Zilin, R., Kajia, C., M., L. M., Kai, W., & Yunyun, Z. (2022). CancerVar: An artificial intelligence–empowered platform for clinical interpretation of somatic mutations in cancer. Science Advances, 8(18), eabj1624. https://doi.org/10.1126/sciadv.abj1624 https://www.chop.edu/news/chop-researchers-develop-new-computational-tool-interpret-clinical-significance-cancer
Learn More:
Top Bioinformatics Books ↗
Learn more to get deeper insights into the field of bioinformatics.
Top Free Online Bioinformatics Courses ↗
Freely available courses to learn each and every aspect of bioinformatics.
Latest Bioinformatics Breakthroughs ↗
Stay updated with the latest discoveries in the field of bioinformatics.
Tanveen Kaur is a consulting intern at CBIRT, currently, she's pursuing post-graduation in Biotechnology from Shoolini University, Himachal Pradesh. Her interests primarily lay in researching the new advancements in the world of biotechnology and bioinformatics, having a dream of being one of the best researchers.











{kind=link}