
The challenge of connecting a protein sequence to its fundamental biological function is becoming more and more significant in the field of biology. The retrieval-augmented protein function prediction method ProtEx, which uses database exemplars to increase accuracy and robustness and allow for generalization to unknown classes, is proposed by researchers from Google DeepMind in this paper. The method used by researchers achieves state-of-the-art results for predicting Enzyme Commission numbers, Gene Ontology words, and Pfam families across numerous datasets and contexts thanks to the fine-tuning strategy used to appropriately condition predictions on retrieved exemplars. In training data, this method works especially well for classes and sequences that are underrepresented.
Introduction
Using ontologies such as Pfam families, Enzyme Commission numbers, and Gene Ontology words, researchers can document the diverse roles that proteins play inside organisms. Protein mapping to functional annotations can help solve important issues in chemistry, biology, and medicine. However, computational protein function predictions are very important since wet lab studies are expensive and protein sequence databases are expanding quickly. The function of a protein can be directly predicted from its amino acid sequence using deep learning techniques, whereas homology-based algorithms match query proteins to annotated sequences.
One of the most significant challenges facing the Pfam database is the generalization of its annotated sequences over sequence space, especially in the dark matter category of proteins. In this complicated sequence space, the discovery of new classes with few annotated sequences has fueled the database’s expansions and presented formidable obstacles for deep learning and homology-based approaches.
Understanding ProtEx
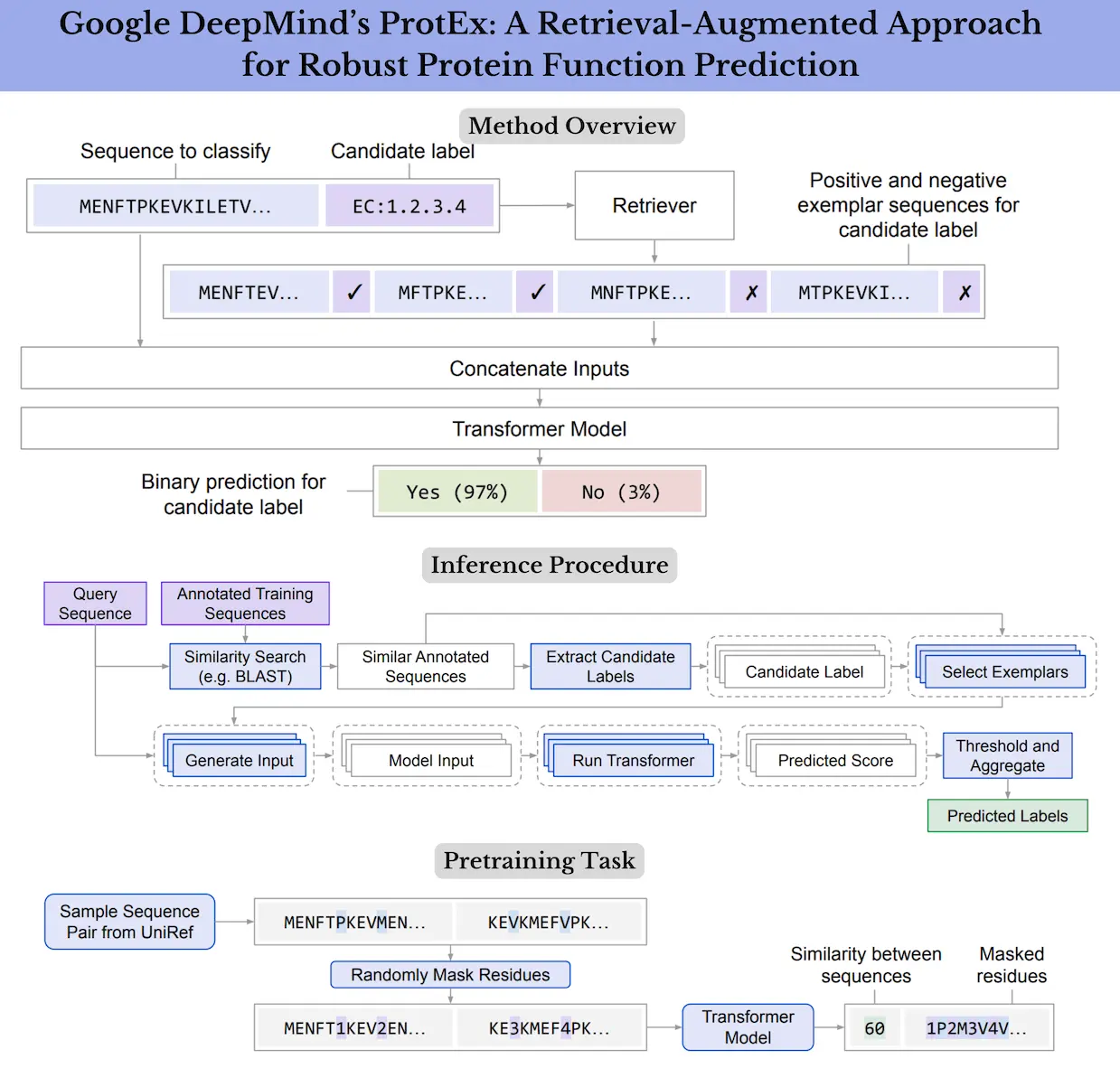
The researchers present ProtEx, a technique for predicting protein functions using recovered exemplars, in this study. To improve accuracy and resilience, ProtEx is a semiparametric technique that blends elements of parametric deep learning models and non-parametric similarity search-based techniques. The retrieval-enhanced approaches in natural language processing and vision, which demonstrate advantages over fully parametric models in capturing tail information and executing few-shot tasks by conditioning on task exemplars, served as the inspiration for ProtEx.
The article shows how to use a homology-based retriever to extract a class-conditioned collection of positive and negative exemplars from the training data using ProtEx, a neural model trained on a query protein and candidate label. The query sequence is then compared to the exemplar sequences by the model to see if it has the same label as the positive exemplars. To effectively learn the relationship between query and exemplar sequences and improve the classification of sequences with low sequence similarity, a novel multi-sequence pretraining objective is proposed.
Why choose ProtEx over BLAST
It can take a while to run BLAST for every example in the training set because the Pfam dataset is much larger than the other datasets. Furthermore, compared to the EC and GO tasks, BLAST does not produce as good of a result for the Pfam classification. Thus, instead of using BLAST, researchers used a different retrieval method that is easier to parallelize and modify. Researchers choose a group of related sequences for each class separately for each sequence. Researchers choose a maximum number of sequences per class at random from the training set for efficiency and then rank these sequences based on a local alignment score that is comparable to the one that BLAST calculates.
Learning more about the Performance of ProtEx
Researchers evaluate ProtEx against two powerful homology-based methods (BLAST and Top pick HMM), a convolutional neural network ensemble called ProtENN, and pre-trained Transformer models called ProtNLM and ProtTNN. Since predicting a binary label without exemplars for the no exemplar ablation performs poorly when there are many classes, researchers adjust the pretrained checkpoint to predict the class label as a string value instead.
Researchers present the average per-family accuracy, which assigns equal weight to all classes, including unusual classes, as well as family accuracy and lifted clan accuracy, which divides families into higher-level clans. The method outperforms the state-of-the-art by a significant amount. Furthermore, ProtEx exhibits more consistent performance across the training set family sizes, with notable increases for cases belonging to uncommon families. In contrast, other approaches often perform worse for examples with rare labels. Additional stratified performance analysis demonstrates that similar trends are seen at the lifted clan level and that the strategy adopted by researchers works effectively even when the sequences have little in common with the closest sequence in the training set.
Limitations
Previous research has demonstrated that the model size and pre-training procedures used in protein inference can be on par with or less than those utilized in the present day. However, because several protein sequences must be encoded and separate predictions must be made, the cost of inference might be higher. The Fusion-in-Decoder (FiD) architecture and candidate label generator are taken into consideration to lessen this. Accuracy and robustness gains could justify the higher processing cost.
Conclusion
ProtEx is a semiparametric method for accurately predicting protein function that blends pre-trained neural models with homology-based similarity search. In this study, researchers employed a general-purpose Transformer architecture and common retrieval techniques (like BLAST) to concentrate on training and inference processes that may successfully condition predictions on retrieved exemplars. Using more sophisticated similarity search methods, like those based on protein structure and more specific structures, the strategy might be further advanced in the future. Lastly, the researcher’s attention was directed to EC, GO, and Pfam label prediction. For future research, other tasks like fitness prediction or creating free-text descriptions of protein function would be interesting.
Article Source: Reference Paper | Codes and model predictions are available on GitHub.
Important Note: bioRxiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Follow Us!
Learn More:




Deotima is a consulting scientific content writing intern at CBIRT. Currently she's pursuing Master's in Bioinformatics at Maulana Abul Kalam Azad University of Technology. As an emerging scientific writer, she is eager to apply her expertise in making intricate scientific concepts comprehensible to individuals from diverse backgrounds. Deotima harbors a particular passion for Structural Bioinformatics and Molecular Dynamics.











{kind=link}