
The authors at Seoul National University developed FoldMason, a free, open-source, progressive Multiple Structural Alignment (MSTA) method published in Science that uses a structural alphabet from Foldseek and aligns hundreds of thousands of protein structures. Achieving two orders of magnitude faster than other methods, when applied to Flaviviridae glycoproteins, it supported phylogenetic analysis below the twilight zone.
Detecting Evolutionary Relationships in Protein Structures
Proteins are central to biology, and comparing them helps us understand how they diverged from common ancestors, how different proteins adapt to different functions, and what particular roles they play in cells. The two approaches to understanding proteins are through their sequences and structures.
While studying proteins through sequences, we compare their linear string of amino acids, which is easy to obtain from genome sequencing. But when proteins diverge too much, sequence similarity drops below the ‘twilight zone’, which is about 20-35% sequence identity, where it becomes challenging to tell whether two proteins are truly related or just similar by chance.
At that point, Multiple Sequence Alignment (MSA) alone can’t reliably detect the evolutionary links. Also, computing the optimal MSA has proven to be NP-complete. This means finding the optimal solution requires checking an enormous number of possibilities, which grows exponentially with the size of the input.
When studying proteins through their structures, we look at their 3D folds, which are more conserved than sequences (as proteins with very different sequences can still fold similarly). This helps us understand function, binding sites, and distant evolutionary relationships better. Multiple Structure Alignment (MSTA) is the process of aligning several protein 3D structures to compare proteins that are evolutionarily related but have diverged too far for sequence-based methods.
MSTA starts from pairwise structure (PSAs) and builds up to multiple alignments. But with millions of predicted structures now available (thanks to AlphaFold and similar tools), manual or small-scale methods are no longer feasible due to the structures being even more complex than sequences. Older methods for MSTA worked well for small datasets but struggled with larger datasets and accuracy. Some alignments even produced false positives, suggesting evolutionary relationships where none exist. Computational tools are sought out to be the only way to handle this scale efficiently.
Building FoldMason on Top of Foldseek
Recent breakthroughs in structure prediction have generated hundreds of millions of protein structures. Databases like AlphaFoldDB (AFDB) and ESMAtlas are massive. Traditional MSTA tools can not scale to the volume efficiently; they would take an impractical amount of time and computing power. Hence, new methods like FoldMason are being developed.
In an earlier study published in Nature Biotechnology, researchers reported Foldseek, a tool that queries large protein structure databases to quickly identify PSAs between proteins. It uses a structural alphabet called 3Di+AA, where 3Di encodes interactions between the residues, and AA encodes amino acid identities. Together, these two transform 3D structures into 1D sequences.
Once structures are represented as strings, researchers can apply fast string comparison algorithms similar to MSA instead of slower 3D superposition calculations. When Foldseek was applied to AlphaFoldDB, it generated AFDB clusters that represent families of proteins that share structural similarities, even if their sequences are very different.
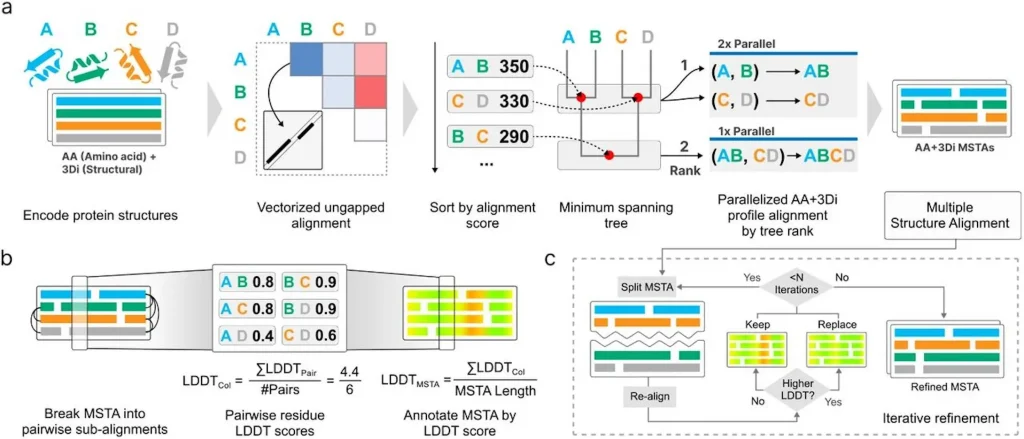
But identifying PSAs is only the first step. To fully understand these clusters, researchers need a fast and scalable MSTA tool to align all members of a cluster simultaneously, suggesting conserved folds, motifs, and evolutionary signals. That’s where FoldMason comes in.
FoldMason builds on Foldseek’s generated structural alphabet approach to perform progressive MSTA efficiently across massive datasets.
Validating FoldMason’s Performance Against Gold Standard Tools
FoldMason was compared to well-known structure and sequence aligners to show that its results are of comparable quality, by curating the Homstrad dataset and AFDB cluster dataset. Homestead is a database of 1032 protein structure families, each having a manually curated reference alignment used as a benchmark for accuracy. AFDB is a large reference-free set of 1000 clusters representing predicted protein structures to test scalability.
Here, FoldMason was used to build evolutionary trees (phylogenies) based on structural alignments of glycoproteins from the Flaviviridae family, which includes viruses like dengue, Zika, and yellow fever. This illustrated its utility in studying viral evolution, where sequence-based methods fail. Structure-based alignments were consistently more accurate, having +6.5% sensitivity, meaning they recovered more correctly aligned residue pairs, and +8.3% specificity, meaning a higher proportion of aligned pairs matched the reference.
Researchers also keep in mind the dynamic nature of proteins and how they adopt different conformations, which can affect MSTA methods. By testing on Calmodulin (CaM), the authors test whether FolaMason struggles when domains shift. Instead, it aligns lobes across both structures, unlike US-align, which relies on rigid superposition. This case study shows that FoldMason is not just fast and scalable but also robust to protein flexibility.
Key Takeaways
The study introduces FoldMason, a novel MSTA tool to overcome the limitations of existing methods. Older structure-based aligners are accurate but slow, while sequence-based aligners are fast but lose accuracy. FoldMason aims to combine structure-level accuracy with sequence-level speed, making large-scale protein structure analysis feasible.
Article Source: bioRxiv | Abstract | Webserver | Github.
Disclaimer:
The research discussed in this article was conducted and published by the authors of the referenced paper. CBIRT has no involvement in the research itself. This article is intended solely to raise awareness about recent developments and does not claim authorship or endorsement of the research.
Follow Us!
Learn More:




Saniya is a graduating Chemistry student at Amity University Mumbai with a strong interest in computational chemistry, cheminformatics, and AI/ML applications in healthcare. She aspires to pursue a career as a researcher, computational chemist, or AI/ML engineer. Through her writing, she aims to make complex scientific concepts accessible to a broad audience and support informed decision-making in healthcare.











{kind=link}