

Comprehensive data atlases have been made possible by the data-driven nature of biomedical research thanks to large-scale single-cell RNA sequencing (scRNA-seq) and spatial transcriptomics (ST). By facilitating a thorough grasp of biology and pathophysiology, these techniques contribute to the identification of novel targets for therapeutic intervention. However, these technologies’ complexity and sheer amount of data provide analytical difficulties, especially when it comes to reliable cell type, integration, and comprehension of intricate spatial interactions between cells. Portrai, Inc. Seoul, researchers created CELLama (Cell Embedding Leverage Language Model Abilities) as a solution to these problems. It is a framework that uses language models to convert cell data into “sentences” that contain gene expressions and metadata, allowing for universal cellular data embedding for a range of analyses. The foundation model CELLama overcomes laborious procedures and manual data selection by providing flexible applications for cell typing and spatial context analysis. Its adaptability is demonstrated by the way its potential alters cellular investigation in a variety of scenarios, from identifying cell types to deciphering complex tissue dynamics.
Introduction
The field of biomedical research has become data-driven with the integration of single-cell RNA-seq and spatial transcriptomics, which has made it possible to create large data atlases in both healthy and diseased states. When paired with comprehensive cellular molecular expression data, these data offer invaluable tools for deciphering pathophysiological mechanisms, identifying novel targets for treatment, and improving our comprehension of disease causes and cellular activities. However, deciphering the spatial arrangements of cells within tissues, analyzing their roles and disturbances, and robustly defining cell types are all made more difficult by the massive amount of data generated. Atlas-level data sets are extensive and complicated, making them difficult to handle with traditional analytics tools. Analytical techniques that make use of vast amounts of atlas-scale data and smoothly integrate with currently held atlas-level information are needed to tackle the issues posed by scRNA-seq and ST analysis for pathophysiology and thorough comprehension of biological events.
Creating foundation models for cellular data has garnered a lot of attention, especially for scRNA-seq and ST data. Transformer architectural models have been developed to incorporate large-scale molecular expression data at the cellular level, including scGPT and Geneformer. Nevertheless, these models are not very flexible in terms of usage or adaption, and they need a lot of training. Their primary focus lies in embedding cells using gene expression data, which may not be appropriate for incorporating additional variables such as spatial context or illness states. Another method uses gene expression ranks and related metadata, including tissue origin, sample condition, data platform, and spatial context, to create “sentences” from cells. By converting cellular data into a language-like structure, this method seeks to give cells a universal embedding space.
Understanding CELLama
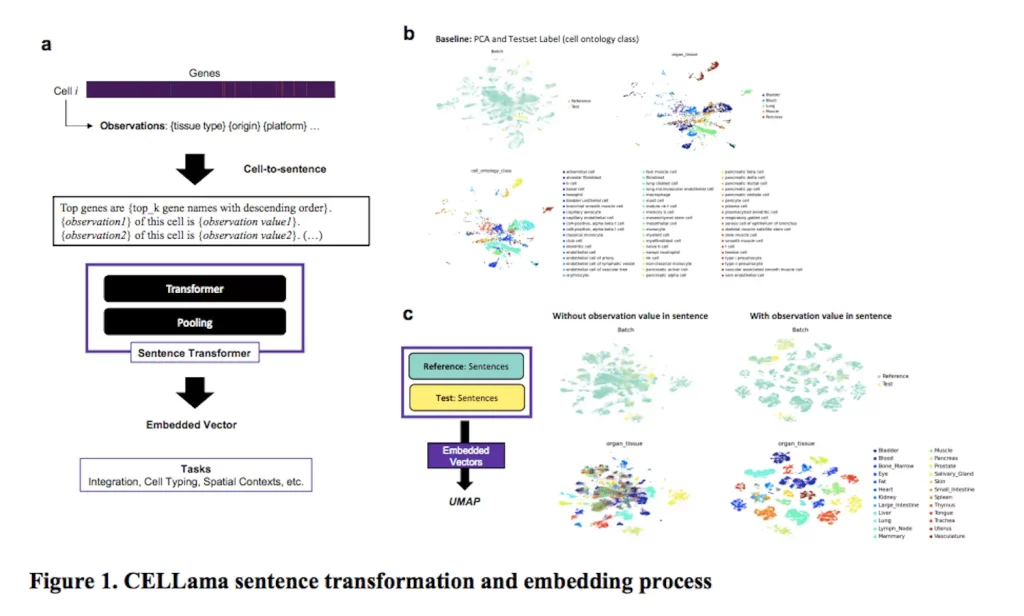
The CELLama framework is intended to be flexible enough to accommodate a wide range of cellular datasets for a variety of general-purpose uses, such as multi-tissue and atlas cell typing. It captures the distinct transcriptome profile of every cell by converting scRNAseq data into sentences in natural language. CELLama can use large-scale cellular data to fine-tune and incorporate pre-trained models. In order to describe the spatial context of cells, information on niche cells can also be incorporated. CELLama pushes the limits of our understanding of complicated biological data by producing phrases from spatial contexts. It exhibits amazing versatility in a variety of applications, from cell type and embedding to spatial context analysis.
Use of Sentence Transformer by CELLama
The machine learning model CELLama uses cell data to generate phrases based on gene expression. The procedure ranks the list of genes according to their expression levels using scRNA-seq data and then sequentially orders the most expressed genes to create a complete phrase. Cell-specific metadata, including the origin of the tissue, the status of the disease, or the experimental situation, are included in this step to improve the sentence generation process. The sentence can also contain important descriptors like tissue type or disease state. The sentences that are produced are a coherent narrative that effectively captures each cell’s expression profile. This narrative is then put into a sentence transformer model that is intended for use with natural language processing (NLP). This model enables efficient training, fine-tuning, and inference procedures of NLP-based packages by flexibly embedding each cell’s data in a high-dimensional space.
Conclusion
By merging large-scale scRNA-seq and ST data into a single data space, CELLama, a data-driven methodology, has completely changed biomedical research. Without depending on manual reference data, CELLama streamlines the cell type process across tissue samples by embedding cellular data into a common space using a language model architecture. This integration also presents a novel method for incorporating cellular metadata, including spatial context, to enable a more sophisticated examination of cellular interactions and behaviors. Transformer-based models are advantageous and scalable in biomedical disciplines due to the flexibility of cell embedding, which is achieved by utilizing natural language processing techniques. As a result of these models’ ability to manage different token lengths and take advantage of neural networks’ long-term memory, it is possible to create flexible phrases from single-cell and spatial data and make them easier to integrate into a common space.
Article Source: Reference Paper | The code for CELLama is available on GitHub.
Important Note: bioRxiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Follow Us!
Learn More:




Deotima is a consulting scientific content writing intern at CBIRT. Currently she's pursuing Master's in Bioinformatics at Maulana Abul Kalam Azad University of Technology. As an emerging scientific writer, she is eager to apply her expertise in making intricate scientific concepts comprehensible to individuals from diverse backgrounds. Deotima harbors a particular passion for Structural Bioinformatics and Molecular Dynamics.











{kind=link}