
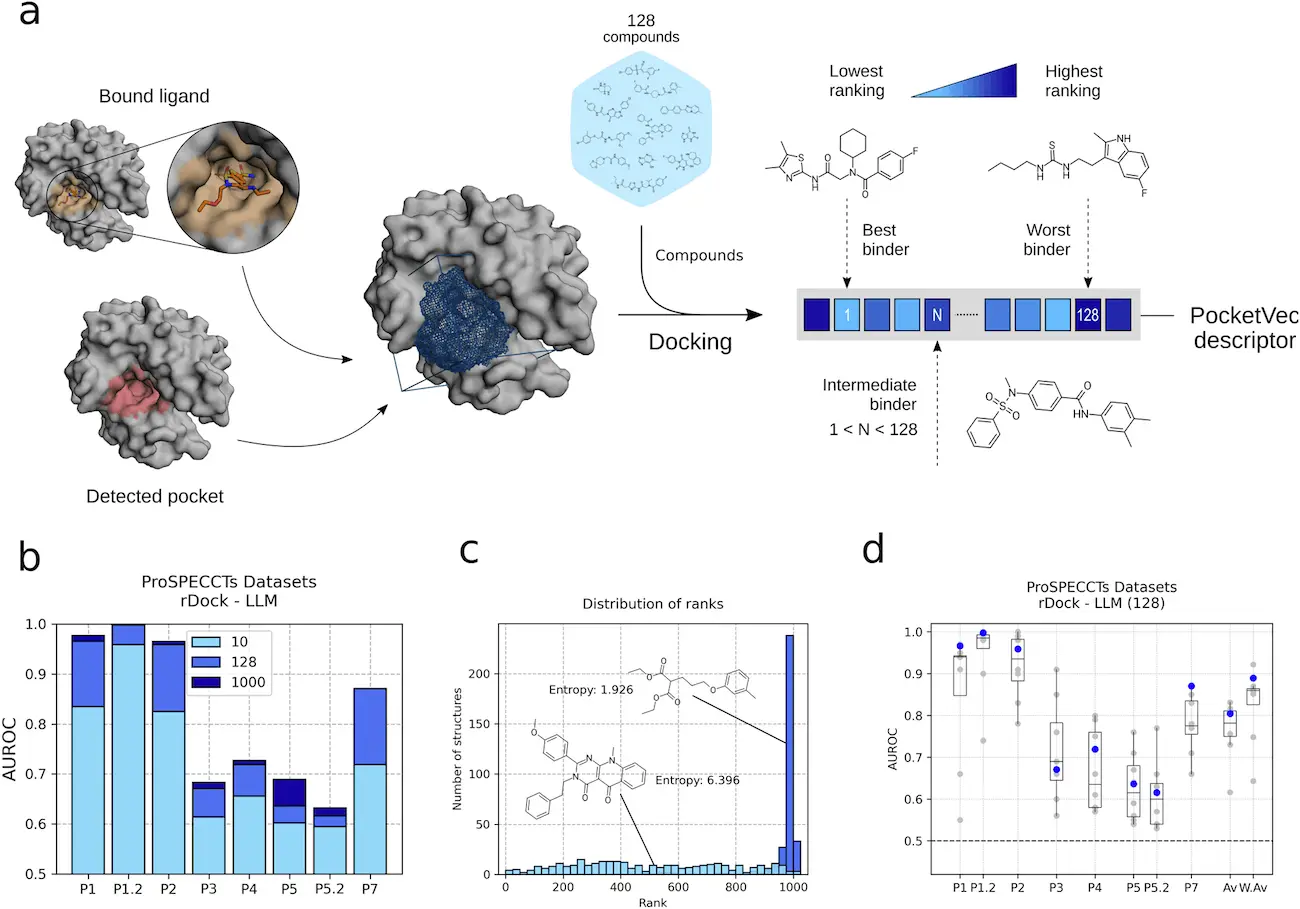
Target-based drug development depends heavily on characterizing druggable pockets, which are protein areas with the capacity to bind organic small molecules. Nevertheless, the process of obtaining pocket descriptors is complex, and the effectiveness of current approaches is frequently restricted. Researchers present PocketVec, a method for creating pocket descriptors by virtually screening lead-like compounds inversely. PocketVec addresses significant issues and performs similarly to leading techniques. Furthermore, researchers employ experimentally established structures and AlphaFold2 models to extensively search the human proteome for druggable pockets, finding over 32,000 binding sites across 20,000 protein domains. Next, researchers create PocketVec descriptors for every site and carry out a comprehensive search for similarities, examining more than 1.2 billion pairwise comparisons. The findings show clusters of similar pockets in proteins lacking crystallized inhibitors and open the door to ways to prioritize chemical probe development to investigate the druggable space. These findings indicate commonalities in druggable pockets not detected by structure- or sequence-based methods.
Introduction
Protein segments known as ligand binding sites engage in biochemical interactions with peptides and other organic small molecules. Eventually, the process of binding causes the protein’s function to be selectively modulated. In fact, one of the most effective methods in traditional drug development is to find tiny compounds that either activate or inhibit a protein linked to a disease based on the high-resolution three-dimensional structure of binding sites.
With the increasing availability of protein structures, the Protein Data Bank (PDB) is a useful tool for the early phases of drug development. Protein-small molecule docking is one example of a structure-based technique that is essential for improving drug design and reducing the risk of clinical trial failure. However, direct comparisons between other proteins and protein families are not possible due to the target-dependent nature of scoring functions. Proteome-wide analyses frequently employ alternative techniques such as interaction fingerprint comparison, binding site similarity assessment, and reverse pharmacophore screening.
Drawbacks of Existing Methods to Generate Pocket Descriptors
- The primary limitation of these methods is that they can only be applied to holographic structures due to the requirement of co-crystallized ligands for the efficient identification of the most significant biophysical interactions taking place in the binding site.
- The handmade nature of the considered binding site representations, which frequently selects parameters based on particular datasets and poorly performs when utilized in more generic and diverse circumstances, is another significant problem associated with pocket descriptors.
- Additionally, several techniques also depend on alignment-dependent comparisons, which makes them especially helpful in offering important insights into the underlying patterns that rationalize binding site similarities. However, this approach also comes with a higher computational cost.
- Furthermore, a well-known issue in the subject is the lack of interpretability of those systems based on deep learning algorithms.
- Lastly, the primary barrier to structure-based drug discovery has long been the lack of access to three-dimensional protein structures, but this is no longer the case.
Making of PocketVec
The limitations of existing pocket descriptors can be overcome by assuming similar pockets bind similar ligands, resulting in similar rankings in a structure-based virtual screening of small molecules. Docking scores tend to correlate more in pockets binding to chemically similar ligands than in pockets binding to dissimilar ligands. This opens the possibility of estimating binding site similarity based on docking rankings and enrichments. Inverse virtual screening, which distinguishes nucleotide and heme-binding sites from a control set of pockets, has been recently applied to distinguish nucleotide and heme-binding sites.
Understanding PocketVec
Here, scientists present PocketVec, a method based on the idea that comparable pockets bind comparable ligands that produce fixed-length, interpretable protein binding site descriptors. The foundation of the process is based on inverse virtual screening, which states that a collection of tiny molecules’ prioritization should be more associated between comparable pockets than between dissimilar ones. Scientists put the strategy into practice and evaluate its accuracy along with the obtained pocket descriptors on multiple pre-established benchmark sets. Additionally, researchers construct PocketVec descriptions for all detected pockets and exhaustively identify drug-binding pockets in experimentally determined and AF2-predicted structures in the human proteome using bound ligands and pocket detection methods.
Conclusion
An innovative PocketVec approach uses inverse docking and the chemogenomics notion that similar pockets bind similar ligands to provide vector-like protein pocket descriptors. It has been applied experimentally to find druggable pockets in the folded human proteome, finding more than 32,000 binding sites spread across more than 20,000 protein domains. After that, an all-against-all pocket similarity search was carried out using PocketVec descriptors, examining more than 1.2 billion pairwise comparisons. This method works in conjunction with other search techniques to find pocket similarities that are missed by comparisons based just on structure or sequence. The study also showed a strong correlation between the likelihood of binding the same chemicals and pocket similarities. All things considered, precise descriptions of druggable pockets could be the basis for the advancement of generative AI methods in drug discovery, providing chances to hasten the creation of a chemical toolkit to investigate biology and, eventually, therapies.
Article Source: Reference Paper | Code availability: GitLab
Follow Us!
Learn More:




Deotima is a consulting scientific content writing intern at CBIRT. Currently she's pursuing Master's in Bioinformatics at Maulana Abul Kalam Azad University of Technology. As an emerging scientific writer, she is eager to apply her expertise in making intricate scientific concepts comprehensible to individuals from diverse backgrounds. Deotima harbors a particular passion for Structural Bioinformatics and Molecular Dynamics.











{kind=link}