
With its recent advancements, scRNA sequencing has enabled scientists to view the biological world at the most local of levels- the single cell. Thanks to scRNA sequencing, we can now examine gene expression in the context of individual cells, enabling us to fathom development, diseases, and the intricate biological networks’ evolutionary cell diversity. The problem lies within scRNA-seq data analysis, which is a very complicated technology for most researchers and often requires advanced programming skills and computational expertise. Enter scExplorer, a user-friendly web server designed to make scRNA-seq data analysis accessible to all. Developed by scientists from Fundación Ciencia y Vida, Santiago, Chile, it opens up new possibilities, allowing researchers to appreciate biology while not being forced to spend time managing computational issues.
Why Did the Researchers Develop scExplorer?
The scExplorer team especially identified a particular bottleneck in the scRNA-seq research. It is worthwhile to note that R’s Seurat or Scanpy in Python have established themselves as powerful platforms for analysis. However, as these tools are programming-dependent, it becomes very difficult for researchers who do not possess a strong computational background to use them effectively. This limitation frequently leaves biologically important questions unasked or hinders advancement toward understanding the dataset’s complexities.
The researchers sought to address this challenge with an approach that would combine the computational power of existing frameworks with ease of use for practical researchers. Work on scExplorer started with the vision of making the scRNA-seq analysis workflow straightforward while allowing for considerable variation in the scale and size of the datasets.
What is scExplorer?
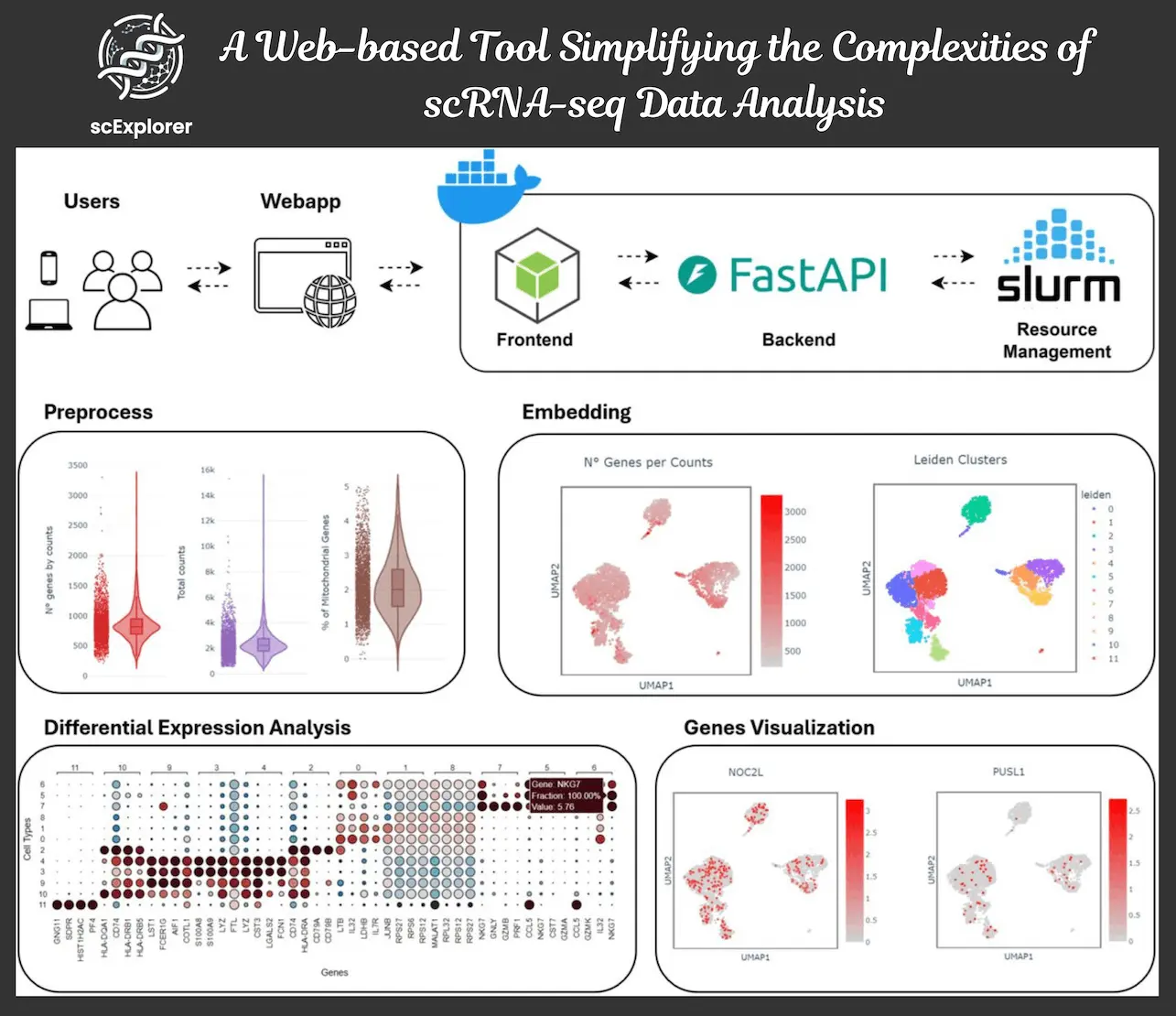
scExplorer is an all-in-one web-based application for the analysis of scRNA-seq data – it includes preprocessing, dimensional reduction, clustering, and differential expression analysis in one package. There are features embedded in the program that do not require any programming expertise to operate. Having been developed on sturdy frameworks including Docker, Node.js, and FastAPI, scExplorer guarantees efficiency in resource utilization and project scalability.
How Does scExplorer Work?
Data Import and Preprocessing
scExplorer supports multiple data formats, including .h5ad, .rds, and 10x Cell Ranger outputs. For data processing, robust filters are applied to identify and exclude low-input cells, genes, and other biological or technical noise components. For instance, it removes cells with high doses of mitochondrial gene expression (a sign of cellular stress) and finds doublets (a pair of cells that have joined into one) using Scrublet for accurate cases of unique cell population representation.
Dimensionality Reduction and Clustering
To interpret the complex high-dimensional single-cell RNA-seq data, scExplorer uses PCA (Principal Component Analysis) followed by UMAP (Uniform Manifold Approximation and Projection) for data visualization. The Leiden algorithm provides the clustering, which also provides users with the opportunity to manually select their interest in one or more cell populations. As a post hoc option, scExplorer includes Clustree which is a visualization interface for optimizing the resolution of the clusters.
Batch Effect Correction
One of the strongest features of scExplorer, and arguably the best of any tool, is how batch effects are addressed and managed. In other words, batch effects are technical differences caused by how a sample was prepared or how a sample was sequenced. The available methods include:
- Combat: Works best when data sets are to be integrated from different sources.
- Scanorama: This is useful in the integration of different populations using their mutual nearest neighbors.
- BKNN: A method that corrects the relationships of data in graph form, but on a more detailed level.
- Harmony: An approach that is modified in such a way that there is coherent biological information despite the existing batch effects.
These methods allow to incorporation of datasets without problems so that problems of nonbiological significance are eliminated and the true essence of the biological problem is studied.
Differential Expression Analysis (DEA)
scExplorer simplifies the identification of marker genes for cell clusters through statistical tests including the Wilcoxon rank-sum test (non-normal distribution) or the t-test (for normal distribution). The results of DEA are shown as visual, interactive graphs and plots that assist the users in quickly selecting the key genes that differentiate the various cell types.
Export and Compatibility
The Intermediate and final outputs including the core outputs can be exported in Python (.h5ad) or R (.rds) formats allowing integration with other subsequent analyses.
Preloaded Datasets and Tutorials
To assist users to start performing functions within scExplorer efficiently, three datasets are preloaded that include the following.
- Human peripheral blood mononuclear cells (PBMCs), serve as a control in scRNA-seq experiments.
- Mouse brain revealing some aspects of neuronal heterogeneity.
- Zebrafish cranial neural crest cells are good for development and lineage tracing.
The comprehensive tutorial helps the users to become familiar with the platform as well as the separate features and parameters of the platform one by one. The availabilities of the preloaded datasets and expertise on how to use the data make it possible even for first-time users to make use of scRNA-seq analysis.
Why Choose scExplorer Over Other Tools?
Other tools can be used, such as CellxGene, ASAP, and Loupe Cell Browser, for specific functions, but scExplorer can be described as being of general purpose. Unlike CellxGene, which is focused on how data is visualized or just data annotation, as is the case for ASAP, scExplorer has a complete order, which is preprocessing and receiving DEA. Its fusion with batch effect correction tools, including those embedded in the R and Python frameworks, provides the much-desired versatility.
ScRNA-seq analysis has been made easier by the introduction of scExplorer as it has eradicated techniques that may be difficult to master. Those with minimal computational expertise, for instance, researchers, can now undertake analyses that were previously complicated, and this opens doors to new findings, especially in developmental and disease biology.
In addition, scExplorer encourages inter-disciplinarity collaboration by enabling biologists, bioinformaticians, and clinicians to perform most tasks effortlessly. The simple but effective interface is complemented by a powerful computational backend to make scExplorer a versatile solution for diverse cellular stakes and genetic target identification easily.
Conclusion
scExplorer is an important development for the scRNA-seq field, as it provides easy yet comprehensive facilities for data analysis. With a distinctive focus on both the elements of graphic design and best software engineering practices, it equips the researchers for efficient data utilization.
It does not matter if you are working on the diversity of immune cells, the development of the brain, or the heterogeneity of tumor tissues — scExplorer has the functionalities to assist you in converting the data into biological knowledge. ScExplorer’s combination of power and ease of use will allow it to become a valuable tool for single-cell experts.
For more information, check out scExplorer and experience how this innovative platform can transform your scRNA-seq analysis workflow!
Article Source: Reference Paper | scExplorer is accessible at Link | The source code is open and freely available on GitHub.
Disclaimer:
The research discussed in this article was conducted and published by the authors of the referenced paper. CBIRT has no involvement in the research itself. This article is intended solely to raise awareness about recent developments and does not claim authorship or endorsement of the research.
Important Note: bioRxiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Follow Us!
Learn More:




Anchal is a consulting scientific writing intern at CBIRT with a passion for bioinformatics and its miracles. She is pursuing an MTech in Bioinformatics from Delhi Technological University, Delhi. Through engaging prose, she invites readers to explore the captivating world of bioinformatics, showcasing its groundbreaking contributions to understanding the mysteries of life. Besides science, she enjoys reading and painting.









{kind=link}