
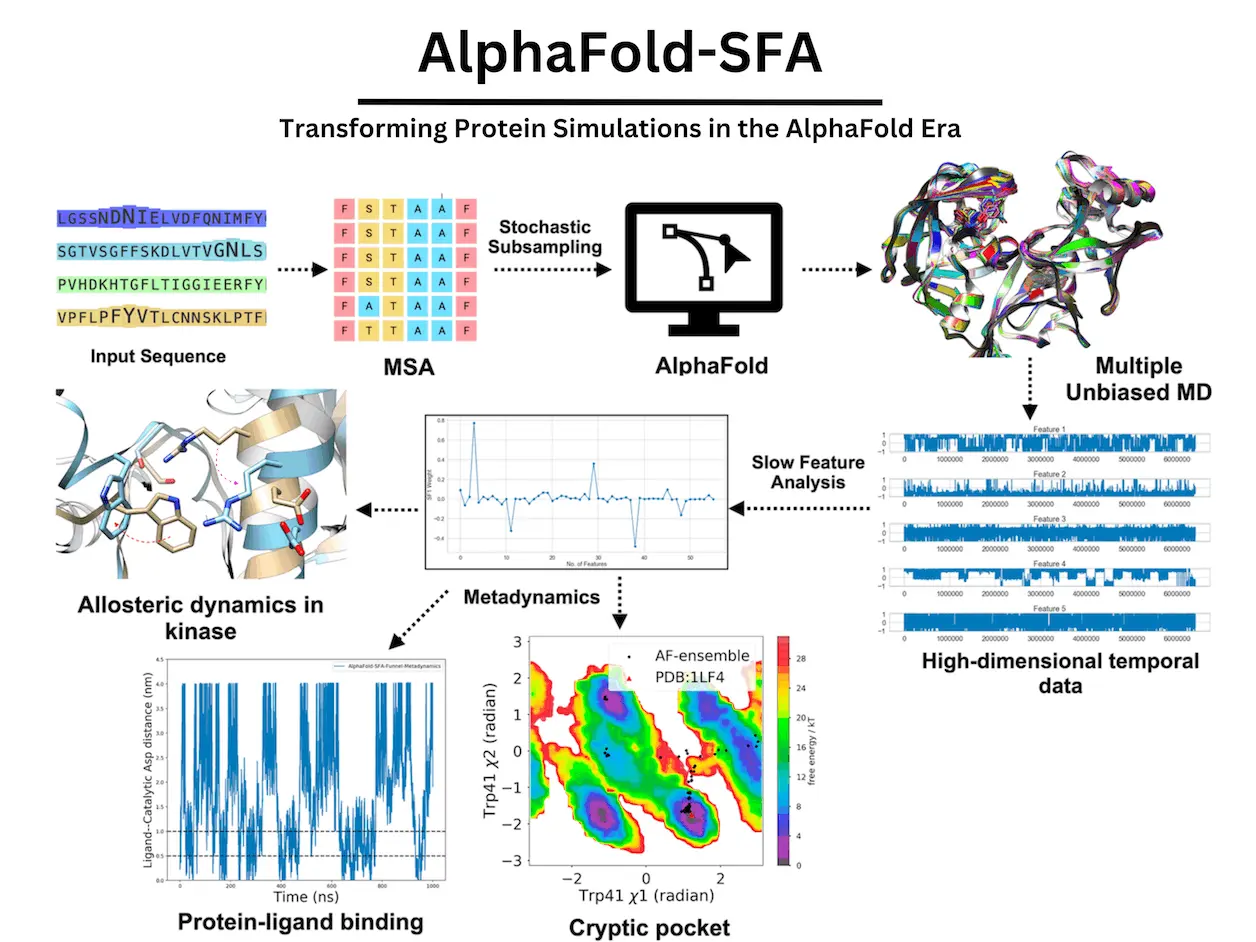
Rare events in proteins, which are essential for comprehending intricate phenomena like transitory structural changes and protein-ligand interactions, are frequently missed by unbiased molecular dynamics. The high free energy barrier separating metastable states is the cause of this comprehension gap in these processes. In this work, a novel unsupervised learning technique called slow feature analysis (SFA) was presented. Its purpose is to extract features with slow variations from high-dimensional temporal data. Protein-ligand binding and cryptic pocket opening are two unusual occurrences that are well-represented by SFA, an unbiased molecular dynamics simulation. Protein conformational plasticity upon ligand binding is captured using metadynamics simulations that sample deep cryptic pocket openings. In order to further structure-based drug discovery, this technology connects the dots between AlphaFold, molecular dynamics simulation, and metadynamics.
Introduction
Protein 3D structure prediction from amino acid sequences has long been a challenge for structural biology. The analysis of complex biomolecules has relied heavily on cryogenic electron microscopy (Cryo-EM) and X-ray crystallography. However, in 2021, an AI-driven approach called AlphaFold transformed the field of protein structure prediction by using protein sequences to predict 3D structures. On the basis of this breakthrough, ColabFold democratized AI-powered protein structure prediction on Google Colab. The conformational dynamics of biologically relevant protein movements, such as cryptic pockets or allosteric communication, are not captured by AlphaFold, X-ray crystallography, or cryo-EM; nonetheless, they are excellent in capturing static representations of proteins. This demonstrates how much more work in structural biology is required to fully comprehend protein structures.
Knowing about Cryptic Pockets
Proteins that are accessible via infrequent conformational changes or interactions with tiny molecules are said to have cryptic pockets, which are secreted binding sites. Because these sites can produce highly selective, powerful inhibitors with distinct structural properties, they are important in the drug discovery process. Sampling apo protein cryptic pocket openings is interesting because these pockets can produce inhibitors against difficult targets like plasmepsin-II, an essential enzyme in the development of antimalarial drugs. Comprehending the concept of allostery in protein kinases is crucial for comprehending the manner in which these enzymes engage with their protein partners and control subsequent signaling in diverse disease pathways.
Aim of the Study
The study investigates the conformational allostery of the immunological and inflammatory response-related serine-threonine kinase RIPK2. In NOD-like receptor pathways, RIPK2 is an essential signaling protein necessary for identifying intracellular pathogens and danger signals. XIAP (X-linked Inhibitor of Apoptosis Protein) mediates the ubiquitination of RIPK2 upon activation, hence influencing downstream signaling pathways, specifically the NF-κB and MAK pathways. Gaining knowledge of RIPK2 allosteric signatures can help with understanding kinase activity and may lead to the discovery of novel therapeutic approaches for a range of pathological disorders.
More about MD Simulation
Rare transitions can be sampled by molecular dynamics simulations, but it can be difficult to capture them in the necessary level of detail due to their restricted timeframes. Stochastic subsampling of multiple sequence alignment (MSA) is a novel technique that has been created to sample a variety of conformational ensembles of three-dimensional protein structures, even in states of high energy. This method can produce a Boltzmann-weighted probability distribution linked to cryptic pocket opening when paired with Markov State Modelling (MSM). However, tens of microseconds of total simulation time are needed to build a robust MSM. An alternative is provided by metadynamics, which deposits collective variables—gaussian-shaped bias—along predetermined reaction coordinates.
Understanding SFA
An unsupervised learning technique called slow feature analysis (SFA) can extract different features from high-dimensional temporal data produced by MD simulations. SFA is employed in metadynamics simulations to capture protein-ligand interaction and cryptic pocket opening in plasmepsin-II, a malaria therapeutic target. Moreover, it records allosteric hotspots linked to protein-protein interaction mediated by receptor-interacting protein kinase 2 (RIPK2), a crucial checkpoint connected to inflammatory reactions. By capturing the Boltzmann distribution associated with a variety of biological events, SFA fills the void between AlphaFold and metadynamics.
Applications of SFA
- AlphaFold-generated structural ensembles were used to train SFA on tiny independent unbiased molecular dynamics simulations, which allowed SFA to catch Trp41 flipping in plasmepsin II, which is required for cryptic pocket opening. A further important aspect that it captured was Tyr77 flipping along the χ1 angle. When Tyr77 and Trp41 are flipped simultaneously, a fully “open” cryptic pocket that is ready for ligand binding is revealed.
- It has been demonstrated that metadynamics utilizing SFA augmented funnel metadynamics can capture a variety of ligand binding events, such as the flap opening and Trp41 flipping. Compared to conventional funnel metadynamics, this method allows for the capture of faster ligand binding. A deep cryptic pocket required for ligand binding was discovered by the simulation employing SFA as CVs, which makes it a novel substitute for route CVs in capturing protein dynamics in plasmepsin II. This strategy is essential for plasmepsin-II ligand binding.
- Using AlphaFold seeded MD simulation as training data, SFA was able to capture crucial conformational fluctuations linked to RIPK2, such as the flipping of Phe165 in the DFG moiety. The first two slow features in the metadynamics simulation were used to catch Phe165 flipping in apo RIPK2, an uncommon occurrence that was not sampled in the ~500 ns unbiased MD simulation.
Conclusion
The study offers a novel way to integrate metadynamics and slow feature analysis (SFA) using AlphaFold seeded molecular dynamics (MD) simulations to examine allosteric dynamics, protein-ligand binding/unbinding, and cryptic pocket opening in biomolecules. Multiple transitions between closed and open states of the cryptic pocket in plasmepsin II were detected in a matter of a few hundred nanoseconds by combining SFA with metadynamics, which greatly improved upon previous techniques. By capturing numerous instances of ligand binding and unbinding, the SFA-augmented funnel metadynamics technique revealed the critical role played by the activation loop and DFG moiety in RIPK2’s transition.
Article Source: Reference Paper | Data Availability: https://osf.io/wm6vx/ | Jupyter notebooks: GitHub | Software: GitHub.
Follow Us!
Learn More:




Deotima is a consulting scientific content writing intern at CBIRT. Currently she's pursuing Master's in Bioinformatics at Maulana Abul Kalam Azad University of Technology. As an emerging scientific writer, she is eager to apply her expertise in making intricate scientific concepts comprehensible to individuals from diverse backgrounds. Deotima harbors a particular passion for Structural Bioinformatics and Molecular Dynamics.











{kind=link}
[…] Unlocking Protein Secrets: AlphaFold-SFA’s Breakthrough in Cryptic Pocket Discovery […]