
Novel compounds can be discovered in the wide chemical space due to generative drug design that enables one to develop molecules that are effective against their pathogenic target proteins. However, designs tend to focus narrowly on certain specific drug-related qualities, thereby limiting the practicality for which created compounds can be used or their abilities to work, thus reducing the success rate in drug development. Addressing such issues, a new approach called TamGen– a framework inspired by a chemical language model similar to GPT- has been introduced by researchers from the University of Science and Technology of China, Microsoft Research AI for Science, and Global Health Drug Discovery Institute, China. The compounds generated by TamGen were of better quality and molecularly viable for drug discovery. The results pave the way for further developments in the area by justifying the application of new methods for drug design.
Introduction
Generative drug design is one promising approach to drug discovery, allowing the generation of new molecules with particular pharmacological properties without dependence on existing templates or molecular scaffolds. This method leads not only to uncovering underexplored classes and unique compounds not coming out of existing libraries but also to the exploration of the vast chemical universe out there, which contains more than ten possible molecules. This is particularly important for target proteins for which no hit compound exists or has developed resistance against currently available therapeutics.
Generative modeling approaches have witnessed an ascent in their application, which is concerned with generating drug-like molecules from target protein profiles. Such methods involve autoregressive models, GANs, VAEs, and diffusion models. All these methods rely on AI tools and demonstrate that target-based generative drug design is feasible with high chances through deep learning. Unfortunately, most of the produced compounds lack desirable properties, including physicochemical properties, such as synthetic accessibility; hence, they often end up not having biophysical or biochemical validation processes. However, it is important to know that these techniques yield a considerable number of new compounds, but all of them are ineffective and inefficient in increasing the actual-world drug development speed.
Understanding TamGen
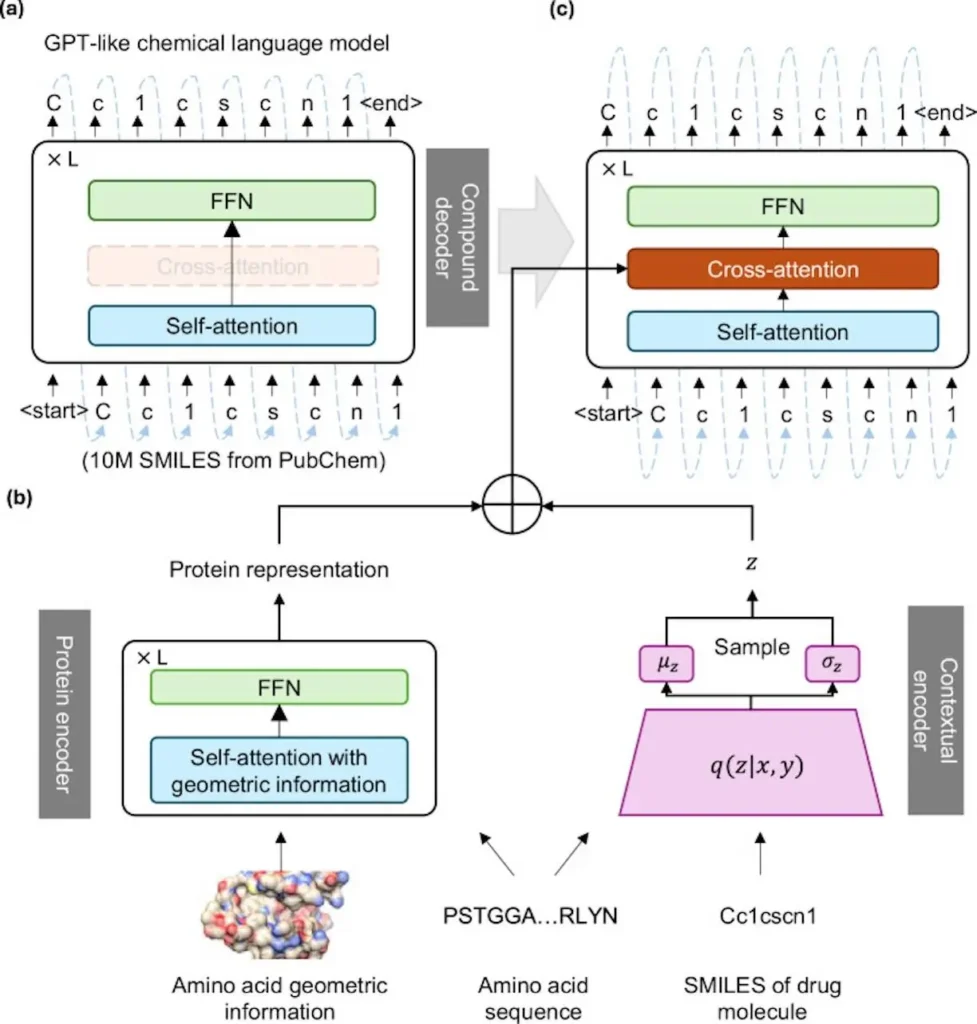
TamGen is a method of molecular generation with target awareness, generating drug-like compounds through a chemistry language similar to GPT. TamGen generates chemical compounds in 1D space with the Generative Pre-trained Transformer (GPT), represented by the Simplified Molecular Input Line Entry System (SMILES), as it draws inspiration from big language models. Modules that encode target protein and compound information to enable target-aware generation based on protein structures and compound refinement based on seeding compounds. TamGen not only produces molecules with greater plausibility, but it also improves the balance between pharmacological activity and synthetic accessibility.
Applications of TamGen
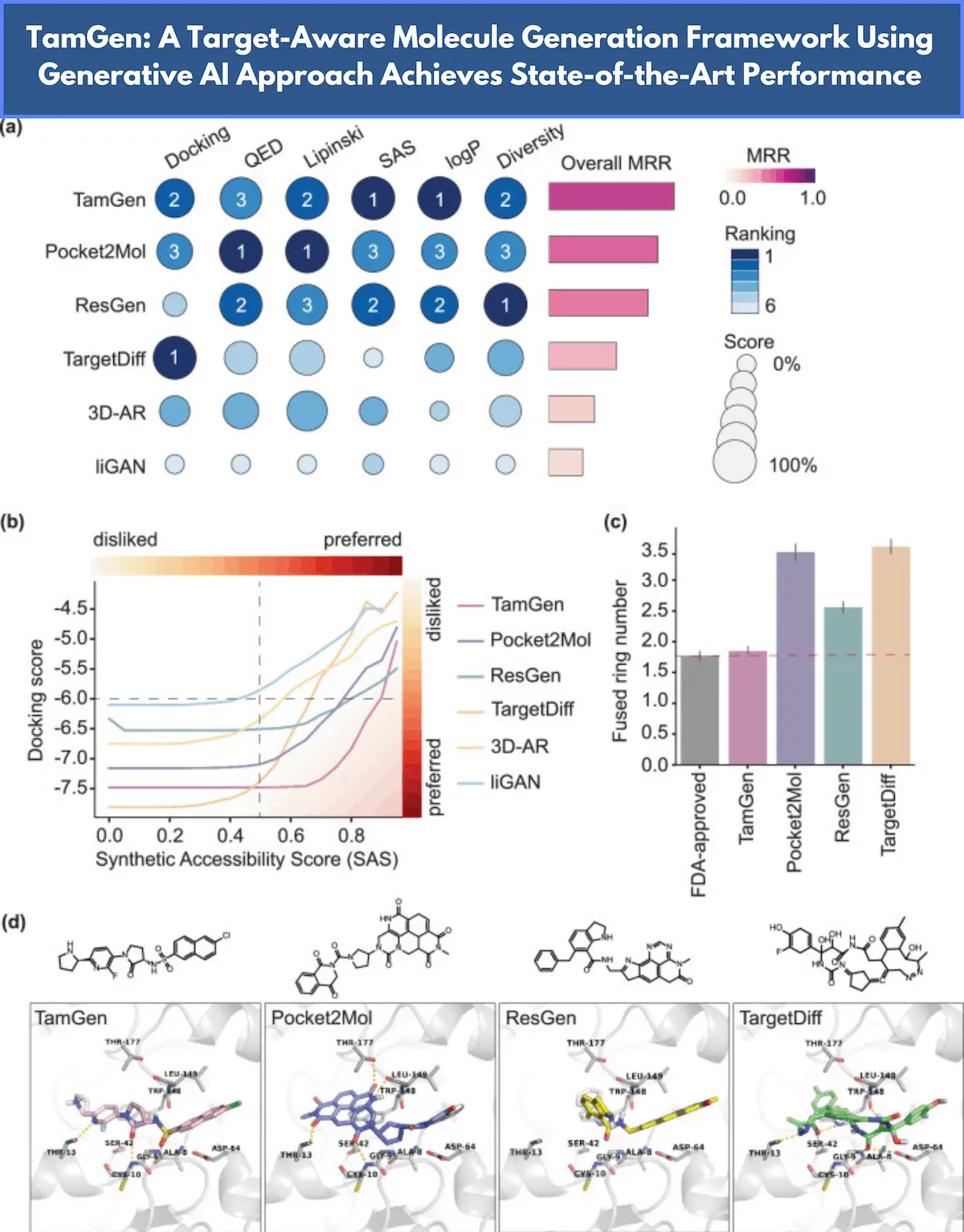
TamGen has produced compounds against the infectious ‘Mycobacterium tuberculosis‘ (TB) that killed 1.3 million people in 2022 and caused 10.6 million new cases that same year. The need for innovation in treating tuberculosis is urgent because of the increasing number of antibiotic-resistant tuberculosis cases found. The study focused on Caseinolytic Protease P (ClpP), a serine protease that is vital to the bacterial protein degradation pathway and is a new target for the development of antibiotics. A TamGen-powered Design-Refine-Test pathway identified 14 candidate compounds significantly active against Mtb ClpP. The diversity of target-aware drug design applications appearing in TamGen is well demonstrated by these compounds that increase candidate pools for optimization and provide useful anchors for hit expansion and structure-activity relationship synthesis.
Factors contributing to successful TamGen applications.
- The pre-trained compound decoder model enables the generation of compounds of high quality and drug development-related properties and compliance with chemical laws.
- Binding pocket modeling incorporates geometric and sequential information, producing highly diverse and drug-like compounds.
- It allows the refinement of candidate molecules of hit compounds through a contextual decoder based on the Variational Autoencoder (VAE). Strong inhibitor compounds against Mtb ClpP were designed thanks to this feature, which allowed for further refining in TamGen and augmenting the probability of better inhibiting compounds.
Limitations of TamGen
- It is poorly sensitive in distinguishing targets that have only subtle differences, such as point mutations or protein isoforms.
- Moreover, understanding the target protein structure and possible binding pockets is essential, as TamGen represents a structural-based drug design method.
Conclusion
Molecules with high binding affinities to target pathogenic proteins are generated via generative AI methods such as TamGen to revolutionize the drug discovery process. Such techniques could facilitate a search for more chemicals than those available in existing compound libraries. TamGen also derailed many compounds against the ClpP protease of Mycobacterium tuberculosis, the line that causes tuberculosis, in what proved to be a state-of-the-art benchmark test. TamGen is a powerful and extremely intuitive tool for structure-based drug design in the sense that it can generate new compounds based on the structural and pocket information of target proteins alone. The versatility of TamGen suggests that it can be effectively employed in treatments for a diverse range of targets as well as in investigating the adaptability of TamGen for use in many biological contexts.
Article Source: Reference Paper | Reference Article | Code is available on GitHub and Zenodo.
Disclaimer:
The research discussed in this article was conducted and published by the authors of the referenced paper. CBIRT has no involvement in the research itself. This article is intended solely to raise awareness about recent developments and does not claim authorship or endorsement of the research.
Follow Us!
Learn More:




Deotima is a consulting scientific content writing intern at CBIRT. Currently she's pursuing Master's in Bioinformatics at Maulana Abul Kalam Azad University of Technology. As an emerging scientific writer, she is eager to apply her expertise in making intricate scientific concepts comprehensible to individuals from diverse backgrounds. Deotima harbors a particular passion for Structural Bioinformatics and Molecular Dynamics.











{kind=link}