
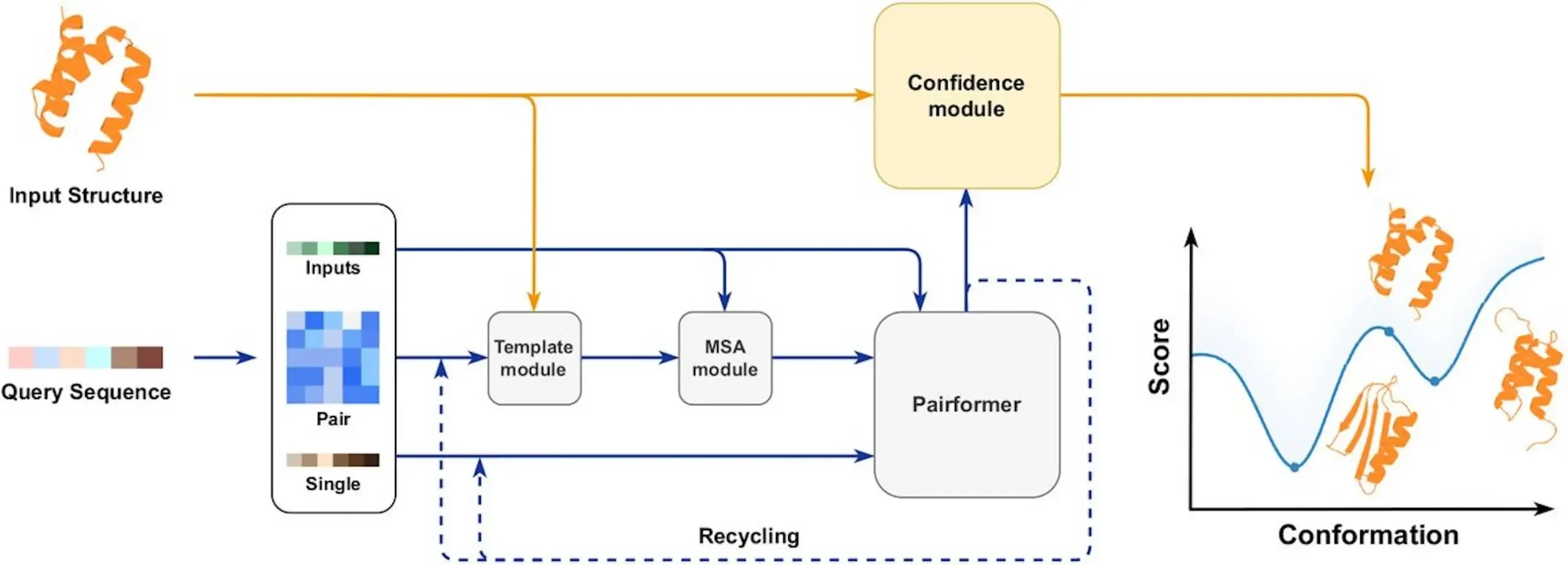
Researchers from Changping Laboratory in Beijing introduced AF3Score, a novel adaptation of AlphaFold3 designed to evaluate biomolecular structures with unprecedented accuracy. This innovation addresses a critical gap in computational biology: the need for reliable, unbiased scoring methods to assess the quality of protein structures, complexes, and designed biomolecules. By reimagining AlphaFold3’s architecture, the team created a tool that could transform how researchers validate and refine molecular models, from drug discovery to understanding dynamic protein behavior.
The Challenge of Biomolecular Scoring
While the 3D structures of proteins and their complexes are the workhorses of biology, they remain a stubbornly persistent problem. Traditional approaches to 3D modeling, such as molecular docking and molecular dynamics simulations, employ energy functions that face difficulties telling apart nearly native structures from decoys. AlphaFold2 solved this problem beautifully by achieving astonishing accuracy in structure prediction, although biases were introduced through its scoring mechanism based on iteratively refining the structure. For instance, while assessing a designed protein binder, AlphaFold2 risks fitting too closely to the model it constructed rather than using the structure provided. Such biases limited the protein’s usefulness for high-throughput screening or analyzing other possible conformations.
In 2024, AlphaFold3 marked a significant turning point in the predictive modeling of biologically relevant structures by integrating proteins with nucleic acids and small molecules into complexes. Despite this groundbreaking step, its structure diffusion-based mechanism still needed to construct new scoring conformations for frameworks that users supplied. The Changping Laboratory team recognized that bypassing this step could unlock AlphaFold3’s latent potential as a universal evaluator.
How AF3Score Works: Simplifying AlphaFold3
By discarding the genetic search and template modules, AF3Score optimizes AlphaFold3 for scoring-specific requirements. Instead, structural coordinates are routed straight to AlphaFold3’s confidence head, which estimates the accuracy of predictions. This skips the new structure diffusion process, allowing AF3Score to assess models without modification.
Picture giving a protein structure to AF3Score: it computes a confidence score based on how coherently the atomic coordinates fit within the neural network’s learned physical and evolutionary frameworks. The score encapsulates the degree to which the structure is plausible from a biophysical standpoint. This strategy avoids the self-reinforcing bias of previous iterative confidence score inflation through self-boosting refinements.
Benchmarking Success: From Proteins to Fold-Switching
The researchers rigorously tested AF3Score across diverse biological systems:
- Monomeric Proteins: Employing the Rosetta decoy dataset, AF3Score achieved a Spearman correlation of 0.834 with TM-scores, surpassing Rosetta (0.760) and equaling DeepAccNet (0.831). Most impressively, it pinpointed near-native structures with an average TM-score of 0.938, competing for rigorously tailored frameworks.
- Protein-Protein Complexes: In the CASP16 Quality Assessment Challenge, AF3Score placed 9th out of 25 methods, illustrating strong performance even for novel interactions outside the training data.
- De Novo Binder Design: In the context of 10 therapeutic targets, AF3Score surpassed state-of-the-art benchmarks RosieTTAFold2 and AlphaFold2 in 8 instances. Pairing it with metrics from AlphaFold2 increased the experimental success rate from 15.2% to 31.6%.
- Fold-Switching Proteins: AF3Score provided equal confidence scores to alternative conformations suggested in proteins such as Adenylate Kinase and KaiB, which allow for more than one functional state, unlike AlphaFold2, which prefers the dominant fold. This approach is vital for the understanding of proteins that can assume multiple functional states.
Practical Applications in Drug Discovery
AF3Score’s most spectacular applications may be in the design of new proteins. For example, creating binders for important targets such as EGFR (a protein associated with cancer) requires both high stability and binding affinity. In the last competition, AF3Score captured 75% of successful binders within its top 1% ranked designs, while AlphaFold2 only captured 25%. When used to filter molecular dynamics trajectories or docking poses, it could accelerate the identification of viable drug candidates.
Also, AF3Score can estimate fold-switching proteins, which makes it possible to analyze more dynamic biological phenomena. Immune evasion by some viral proteins often involves the exploitation of conformational changes. By scoring alternative states, researchers can design inhibitors that stabilize dihedral angles of less pathogenic states.
Conclusion
The repurposing of AlphaFold3 led to the creation of AF3Score, which automates the evaluation of biomolecular structures, showcasing how model adaptation can transform scientific exploration. By combining precision and efficiency, the Changping Laboratory team created a generic evaluator built on the AlphaFold3 architecture. From drug design catalysis to protein dynamics, the repertoire of biomolecular systems tools—to be understood, one structure at a time—deepens. With ongoing advancements in AI and structural biology, tools like AF3Score will undoubtedly play a pivotal role in the next decade of biomedical discovery.
Article Source: Reference Paper | Code Availability: The code is available on GitHub.
Disclaimer:
The research discussed in this article was conducted and published by the authors of the referenced paper. CBIRT has no involvement in the research itself. This article is intended solely to raise awareness about recent developments and does not claim authorship or endorsement of the research.
Important Note: bioRxiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Follow Us!
Learn More:




Anchal is a consulting scientific writing intern at CBIRT with a passion for bioinformatics and its miracles. She is pursuing an MTech in Bioinformatics from Delhi Technological University, Delhi. Through engaging prose, she invites readers to explore the captivating world of bioinformatics, showcasing its groundbreaking contributions to understanding the mysteries of life. Besides science, she enjoys reading and painting.











{kind=link}