
The ability to anticipate the structure of proteins is essential for improving biological research, developing new pharmaceuticals, and designing experiments. It also helps to comprehend the structure-function link of proteins. Protein structures are dynamic, although this fact has not gotten much attention despite the fact that deep learning techniques and the increased availability of experimental 3D protein structures have sped up the process of structure prediction. To understand dynamic protein structures, researchers from Fudan University and collaborators presented a novel 4D diffusion model using molecular dynamics (MD) simulation data. Combining a unified diffusion model, the model generates dynamic protein structures, including side chains and the backbone. Side-chain dihedral angle predictions and atomic grouping are used to improve structural consistency. A motion alignment module enhances temporal structural coherence across several time steps, and a reference network integrates latent embeddings of original 3D protein structures. This is the first model based on diffusion that can simultaneously predict protein trajectories over several time steps.
Introduction
The intricate connection between the biological function of proteins and their structures is essential for the progress of both biological research and pharmacological development. A protein’s biological functioning is determined by the 3D architecture encoded in its linear 1D amino acid sequence. In computational biophysics, comprehending protein folding has proven to be a major issue. Identifying appropriate templates, their refinement to resemble the native state, improving force field precision and conformational exploration, and efficiently managing the computational costs associated with protein structure prediction are important challenges. These challenges are particularly relevant in free modeling scenarios requiring de novo structure generation.
Understanding MD Simulations
Molecular dynamics (MD) simulations offer a dynamic viewpoint on molecular systems, making them indispensable tools in computational biology. Although they produce excellent data for data-driven techniques, their computing cost frequently increases cubically with the number of electronic degrees of freedom. Deep learning techniques have been used to overcome these constraints; however, the current approaches are mostly useful for proteins with fewer atoms. In this work, dynamic structures of proteins containing hundreds or even thousands of amino acids will be generated by utilizing MD data, including complicated structures with full side-chain representations. By using this method, MD simulations may now be used for increasingly complex and large-scale protein systems, which improves the comprehension of their dynamic behaviors.
Diving Into Deep Learning Techniques
Learning-based structural studies have been substantially expedited by recent developments in deep learning techniques and the exponential proliferation of experimental protein structures in the Protein Data Bank (PDB). With accuracy on par with experimental techniques, AlphaFold2 has brought a revolutionary approach to 3D protein structure prediction. RoseTTAFold uses a three-track network architecture to improve its predictive power, and ESMFold and OmegaFold use high-capacity transformer language models trained on evolutionary data. Research on protein conformation sampling has evolved thanks to large-scale data archives, which produce a variety of structural conformations. Building upon its predecessor, AlphaFold3 incorporates joint structures across proteins, nucleic acids, small molecules, ions, and changed residues through an upgraded computational architecture and diffusion network. Nevertheless, research on dynamic protein architectures is still in its infancy.
Key Features of the Work
This work proposes a novel method for using a 4D diffusion model to simulate dynamic protein structures. Three main areas of focus include the following:
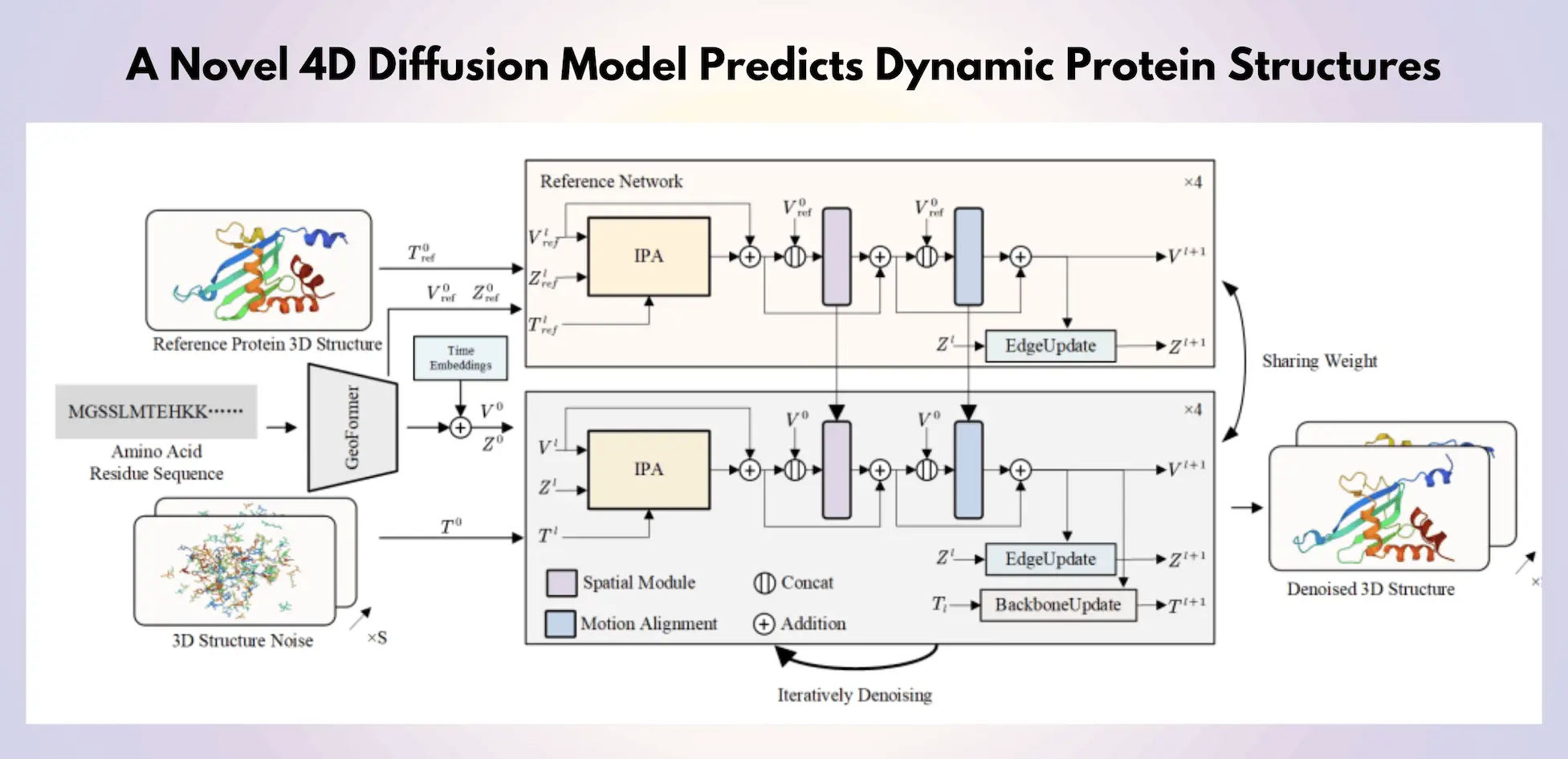
- Researchers first suggest a single diffusion model to predict protein architectures, including side-chain and backbone components. In structures with hundreds of residues, the framework effectively replicates protein motion by grouping atoms within each residue into stiff groups to reduce the degrees of freedom. In order to precisely generate proteins, the diffusion model is guided by node and edge properties obtained from structure prediction models, which reflect the amino acid sequence. To recover individual atomic coordinates based on dihedral angles reliably, researchers employ side-chain dihedral angle predictions and an amino acid atomic model, in contrast to techniques limited to de novo structure prediction.
- Second, to incorporate pertinent features into the denoising diffusion network, the original 3D protein structure is integrated as a condition and recorded through a reference network for latent embedding. The reference network plays a crucial role in preserving the structural integrity of proteins when they move.
- Thirdly, researchers suggest a motion alignment module inside the score-based diffusion network to collect kinetic information from neighboring frames in the diffusion model. This module consists of temporal attention layers. This improvement reduces abrupt transitions during motion by improving the coherence of motion in dynamic proteins that are created.
About 4D Diffusion Model
The diffusion model efficiently produces dynamic protein structures at several time steps, improving productivity and guaranteeing stable sequences in a temporal context. This methodology enhances the synthesis of dynamic protein structures while maintaining coherent and temporally consistent sequences. Dynamic protein structure predictions were obtained for sequences of up to 256 amino acids over 32 time steps by a thorough investigation that used benchmark datasets such as ATLAS and Fast-Folding. This feature makes it possible to describe dynamic protein conformations at different time scales, capturing both notable inter-conformational changes and intra-conformational motions. These results mark a major breakthrough in the prediction of dynamic protein structures.
Conclusion
This work presents a 4D diffusion model that can effectively produce dynamic protein structures at several time steps at once. The unified diffusion model generates protein architectures, including both the main chain and side chains. Researchers also present a motion alignment module that improves sequential coherence in the synthesized dynamic proteins, hence minimizing abrupt transitions during the motion, and a reference network that guarantees the structural consistency of proteins throughout the motion. The system can capture viable conformational changes and explore long-term trajectories through training on the ATLAS and Fast-folding protein datasets, resulting in reliable predictions.
Article Source: Reference Paper
Follow Us!
Learn More:




Deotima is a consulting scientific content writing intern at CBIRT. Currently she's pursuing Master's in Bioinformatics at Maulana Abul Kalam Azad University of Technology. As an emerging scientific writer, she is eager to apply her expertise in making intricate scientific concepts comprehensible to individuals from diverse backgrounds. Deotima harbors a particular passion for Structural Bioinformatics and Molecular Dynamics.











{kind=link}