
Recently, a paper published on Cell reports the EN-TEx, a system/project that challenges conventional functional genomics research by providing a comprehensive resource that goes beyond the use of a single haploid reference genome. The resource contains information on personal epigenomes and associated models that could be combined with other genome annotation resources. By utilizing the diploid genome, this resource is vital for future functional genomics and has the potential to transform precision medicine!
The Need for Improving Functional Genomics Research
Studying genetic variants and their impact on molecular endophenotypes – such as epigenetic activity, RNA expression, or protein levels – is fundamental as it helps us understand the underlying mechanisms of complex biological processes, including diseases.
Many previous research projects like genome-wide association studies (GWAS), expression quantitative trait loci (eQTL) studies like the genotype-tissue expression (GTEx) project, and the ENCODE project have contributed to assessing the impact of genetic variation on molecular endophenotypes, relating to functional genomics research.
However, these studies used the generic haploid reference genome sequenced 20 years ago as part of the Human Genome Project. This is a significant drawback because using a single haploid reference genome means that the genomic sequence represents only a single set of chromosomes from a single individual instead of reflecting the genetic variation existing in a population or of a particular individual of interest. More importantly, heterozygous genetic variations could have different functional outcomes and provide more information about functionality than merely identifying variations using a single haploid reference genome.
Therefore, directly using the variations observed in an individual’s diploid sequence would provide far more accurate information about the molecular endophenotypes and their association with disease phenotype. This is because using a diploid genome means that the heterozygous loci can differentiate sequences from each of the two parental chromosomes (haplotypes). These different sequences give rise to different molecular outcomes from each, such as differential RNA expression of transcription factor binding. In this manner, allele-specific (AS) variants can be identified because the imbalance of expression or epigenetic activity between the haplotypes can be accurately measured by taking the reference allele as a baseline, avoiding biological and technical biases.
The Principles of the EN-TEx Resource
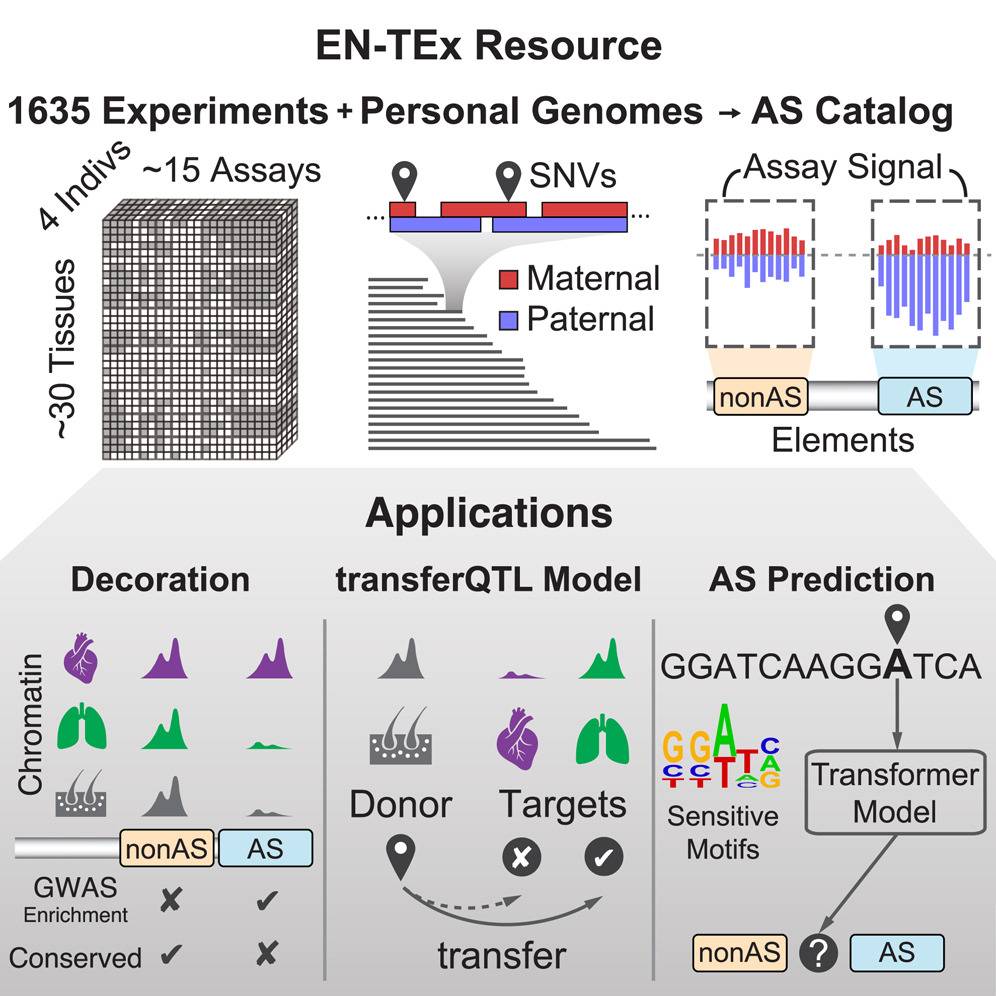
Researchers recently developed the EN-TEx resource, which addresses the issue of relying on a single haploid reference genome in functional genomics research. EN-TEx offers a uniformly processed dataset of roughly 15 functional genomic assays collected from four individuals for approximately 30 different tissues, many of which were difficult to obtain. The dataset contains information on structural variants (SVs) and single-nucleotide variants (SNVs) and also allows for more precise and reliable quantification of differential expression and regulatory-element activity than was previously possible.
One crucial benefit of this resource is that it enables scientists to visualize the influence of genomic variation on chromatin directly and to more accurately quantify the imbalance of RNA expression and epigenetic activity between the haplotypes of a heterozygous genetic variant. This helps distinguish the ‘causal’ AS variants from the AS variants that are only thought to be linked to observed functional changes.
Furthermore, given the uniform nature and large scale of EN-TEx, it enables the creation of the most extensive catalog of non-coding AS variants, which can be leveraged to build generalized models of variant impact. Specifically, the researchers created a fascinating model to predict the allele-specific imbalance resulting from an SNV from the extended local sequence within only a 250 bp window, illustrating the importance of AS-sensitive transcription factor motifs.
Additionally, the resource can be related to external genome annotations, including eQTLs and regulatory elements already known for the human genome. This means that the EN-TEx resource contains information about the epigenome of different tissues, which is used to study gene regulation and variants that affect gene expression. The uniform collection of epigenetic data allows researchers to transfer information about variants affecting gene expression from one tissue to another!
The EN-TEx Project
The researchers used various sequencing technologies to sequence the genomes of the 4 GTEx individuals, determining the diploid personal genomes for each individual. Over 1600 experiments were carried out using 15 assays on 30 tissues. The key finding was that personal genomes significantly impacted gene expression levels and candidate cis-regulatory elements. Also, it indicates that EN-TEx provides a preferred platform to reliably and consistently measure inter-individual, inter-tissue, and inter-assay variability.
Then, by investigating AS behavior on a large scale using EN-TEx, researchers found roughly 800 AS events at SNVs on average in each sample (on 1000 samples). Furthermore, AS SNVs within a genomic element together were also grouped, and on average, 200 ‘AS elements’ were identified in each sample. AS SNVs across all tissues, individuals, and assays were combined, and 1.3 million AS SNVs were identified, representing the AS catalog. The AS catalog covers 76% of common AS SNVs in the European population, including rare SNVs and known pathogenic and deleterious variants. Therefore, given its large size, this AS catalog can be leveraged to determine AS SNVs in an entirely new sample.
The study then demonstrates three broad applications of the EN-TEx
- Decorating ENCODE elements with EN-TEx tissue and AS information:
- Relating AS SNVs to GTEx eQTLs and modeling eQTLs in hard-to-obtain tissues
- Modeling AS activity from the variant impact on the nucleotide sequence, highlighting sensitive TF motifs.
These applications, taken together, focus on the predictive modeling of AS behavior and approaches to decorating existing genome annotations.
Implications and Limitations of EN-TEx and Conclusion
The EN-TEx project has generated a database of personal epigenomes and associated models that can be effortlessly connected with other genomic resources. The impacts of the variants were studied, leading to the crucial finding that the local sequence around a variant is the most significant factor in determining its functional impact. The diploid genome is crucial for accurate and reliable gene expression and regulatory activity analyses. In the future, one can expect personal genome sequences to be generated for every functional genomics experiment, and the EN-TEx personalized epigenetic approach will gain popularity, potentially providing advantages for precision medicine.
Despite the significant findings, the study had certain limitations. The major limitation is that only four individuals were profiled, limiting the study’s statistical power. However, it should be noted that the EN-TEx is designed to be easily scaled up to larger cohorts. Scaling up would allow gaining information about rare variants that are not accessible through conventional QTL studies focusing on genetic variants. Therefore, scaling up to a larger cohort would provide further valuable information and is something that should be undertaken in the future.
Conclusion
To conclude, the EN-TEx project transforms our understanding of human functional genomics and has great potential for future research. The resource contains information about personalized epigenomes and associated models, allowing for a more accurate and reliable measure of differential expression and regulatory activity. It also provides insights and benefits into the idea of scaling up cohorts, potentially transforming precision medicine.
Article Source: Reference Paper
Learn More:




Diyan Jain is a second-year undergraduate majoring in Biotechnology at Imperial College, London, and currently interning as a scientific content writer at CBIRT. His passion for writing and science has led him to pursue this opportunity to communicate cutting-edge research and discoveries engagingly to a broader public. Diyan is also working on a personal research project to evaluate the potential for genome sequencing studies and GWAS to identify disease likelihood and determine personalized treatments. With his fascination for bioinformatics and science communication, he is committed to delivering high-quality content a CBIRT.





{kind=link}