
AlphaPulldown version 2.0, a noteworthy computational biology program that transforms protein structure modeling, is presented by researchers from the European Molecular Biology Laboratory, Hamburg, and the University of Basel. It offers an automated Snakemake pipeline, advanced data compression techniques, and expanded support for multiple modeling backends. This versatile tool can predict protein interactions efficiently, making it crucial for drug discovery, biotechnology, and disease understanding.
AlphaPulldown, introduced earlier, has advantages but still faces challenges in automation, code flexibility, and data handling. AlphaPulldown2 simplifies protein structural modeling through data management optimization for large-scale applications, code flexibility enhancement, and workflow automation. Along with several other enhancements, it offers compressed data storage, an automated Snakemake process, and support for more modeling backends like UniFold and AlphaLink2. Because of these improvements, AlphaPulldown2 is now a flexible platform that can be used to predict complex multi-unit assemblies and binary interactions.
Introduction
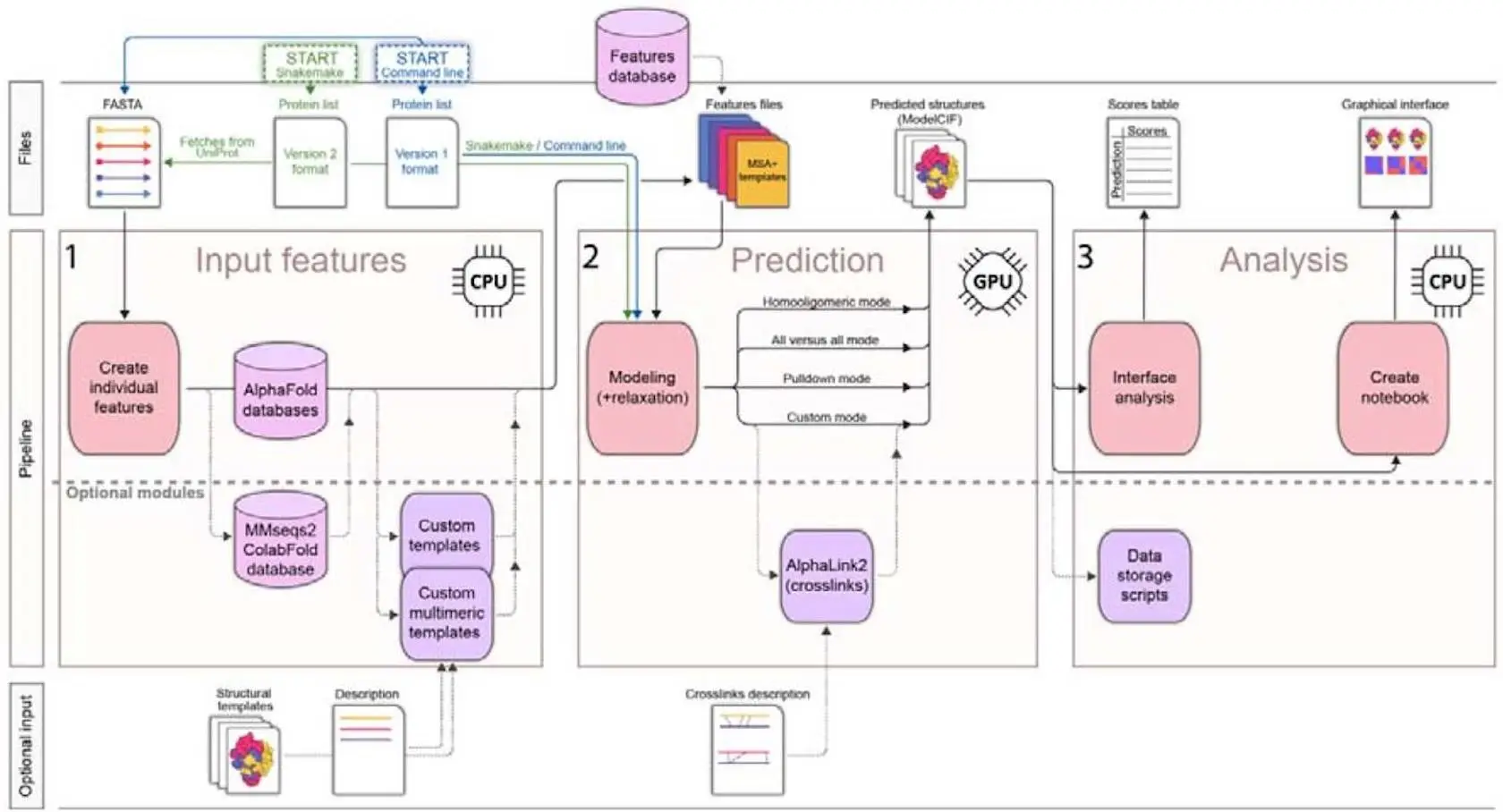
Researchers are now much better able to predict protein-protein interactions (PPIs) and the structure of protein complexes because of recent developments in artificial intelligence (AI). The purpose of the Python package AlphaPulldown was to simplify PPI screens and enable high-throughput modeling of complexes of higher order using AlphaFold-Multimer. By separating the AlphaFold pipeline into GPU-based structure prediction and CPU-based input feature calculation, AlphaPulldown shortens the computation time. It has four modes: custom, homo-oligomer, all-versus-all, and pulldown.
While the all-versus-all mode predicts pairwise PPIs between all proteins in a given list, the pulldown mode filters interactions between one or more “bait” proteins and a list of possibilities. Alternative oligomeric states are modeled by the homo-oligomer mode, while the custom mode permits a variety of protein or fragment input combinations. With the help of an integrated analysis pipeline, AlphaPulldown generates a graphical summary in a Jupyter notebook for a thorough examination of model confidence and interaction properties. This pipeline adds additional evaluation metrics, such as pDockQ and physical interface parameters, to native AlphaFold scores. Numerous applications have made use of AlphaPulldown, which has proven useful for PPI displays, individual complicated modeling, and interface grading.
Looking into AlphaPulldown
A comprehensive tool for predicting protein structures, AlphaPulldown 2.0 has enhanced modeling capabilities and a better user experience. To overcome the challenge of managing numerous jobs and computing resources, it provides a suite for binary PPIs and multi-unit assemblies. In order to ensure a scalable and repeatable pipeline for large-scale structural modeling, AI-based structural modeling is automated using the Snakemake workflow management system.
The original AlphaPulldown method is replicated in this pipeline, but all phases are now carried out automatically in response to an initial setting. Singularity containers are used internally by Snakemake to guarantee repeatability and interoperability with different computation architectures, such as local clusters or the cloud. It resumes from saved checkpoints and reschedules jobs with settings modified according to failure causes. Additionally, Snakemake makes the installation procedure easier because AlphaPulldown is now accessible on DockerHub and is installed immediately on the first run.
A versatile format for configuring modeling jobs has been introduced by the new Python program AlphaPulldown2. All of the AlphaPulldown modes are combined into one simplified format in this format. Additionally, it makes UniProt IDs for input proteins possible, which makes it easier to automatically retrieve sequences. This improves the automation and usability of the Snakemake pipeline while streamlining input preparation and demystifying intricate modeling settings.
Features of AlphaPulldown2
- The AlphaPulldown codebase has been reorganized to take into account feature requests and community interest. Although AlphaPulldown was first developed using AlphaFoldMultimer, additional implementations, such as OpenFold and UniFold, have since surfaced. New tools like HelixFold3, OpenFold Multimer, and AlphaFold3 are coming out. AlphaPulldown2’s architecture has been restructured to enable flexible integration of many modeling backends, such as AlphaLink2 and UniFold. Code restructuring, automated testing, and pipelines for continuous integration and delivery are examples of improvements.
- The main obstacle in structural modeling is ensuring data is accessible and reproducible in a consistent format. The modeling process is thoroughly documented using the ModelCIF format, which is an extension of the PDBx/mmCIF dictionary and conforms to FAIR principles. Model deposition and adherence to FAIR standards are facilitated by this modification, which guarantees AlphaPulldown2-generated models are compatible with repositories such as ModelArchive.
- XL-MS, or cross-linking mass spectrometry, is included in the workflow of the software framework AlphaPulldown2. Cross-linked residue pairs are found for structural modeling using this method, which aids in studying protein and macromolecular complexes.
Future Direction
Researchers intend to include other modeling backends in the future, such as OpenFold, after its multimeric modeling mode is completely functional. As permissively licensed copies of AlphaFold3 become available, researchers will continue to assess and incorporate them, and they are also working on broadening the scope of scoring functions. Future advancements will also concentrate on incorporating more molecules and post-translational alterations made possible by these new modeling backends.
Conclusion
A flexible option for protein complex predictions, AlphaPulldown is a platform for high-throughput AI-based structural modeling that may be customized. Modeling quality is improved by its automation features, compatibility with various modeling backends, and incorporation of experimental data such as cross-links. AlphaPulldown is sustainable for large-scale projects due to its improvements in computing efficiency and storage management, saving time and resources. The more recent version, AlphaFold3, needs more testing before being included in processes such as AlphaPulldown. However, AlphaFold3 has been demonstrated to hallucinate fake secondary structures more frequently than AlphaFold2, and its accuracy has only marginally increased. It continues to be a dependable tool for high-throughput protein complex modeling even though it depends on AlphaPulldown and AlphaFold-Multimer.
Article Source: Reference Paper | AlphaPulldown2 is freely available on GitHub.
Disclaimer:
The research discussed in this article was conducted and published by the authors of the referenced paper. CBIRT has no involvement in the research itself. This article is intended solely to raise awareness about recent developments and does not claim authorship or endorsement of the research.
Important Note: bioRxiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Follow Us!
Learn More:




Deotima is a consulting scientific content writing intern at CBIRT. Currently she's pursuing Master's in Bioinformatics at Maulana Abul Kalam Azad University of Technology. As an emerging scientific writer, she is eager to apply her expertise in making intricate scientific concepts comprehensible to individuals from diverse backgrounds. Deotima harbors a particular passion for Structural Bioinformatics and Molecular Dynamics.





{kind=link}