
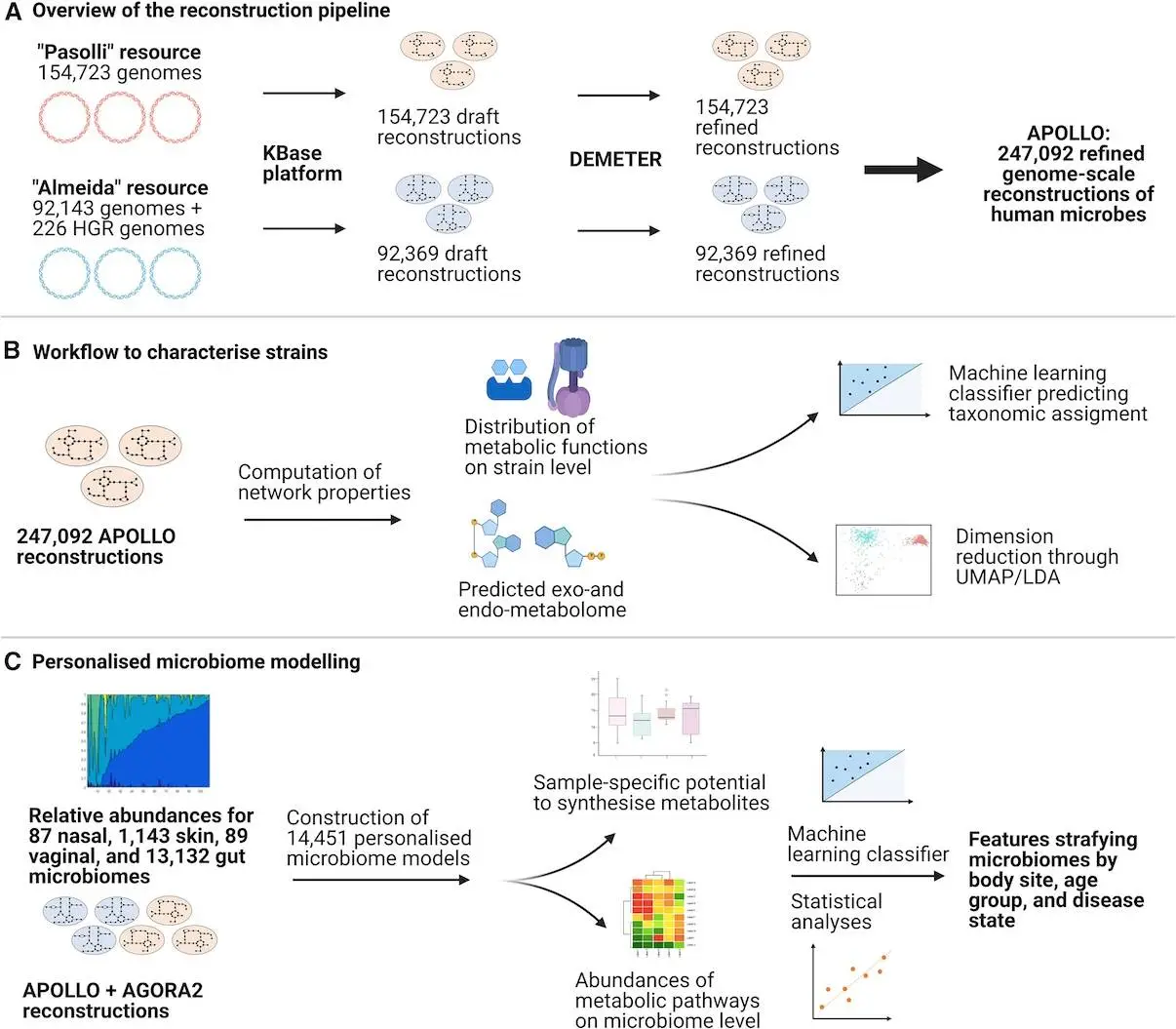
The simulation of diet-host-microbiome-disease relationships is made possible by genome-scale modeling of microbiome metabolism. However, the extent of available genome-scale reconstruction resources is constrained by computational difficulties. To create a database of 247,092 microbial genome-scale metabolic reconstructions encompassing 19 phyla and including more than 60% of uncharacterized strains, the analysis methodology known as APOLLO was created by researchers from the University of Galway, the University of Lorraine, and the University of Padova. Based on calculated metabolic characteristics, APOLLO predicts the taxonomic identification of strains using machine learning. In order to thoroughly examine their metabolic capacities at the community level, it also constructs 14,451 metagenomic sample-specific microbiome community models. The metabolic capacities of uncultured and unclassified species may be systematically examined thanks to this pipeline, which offers hitherto unheard-of possibilities for systems-level modeling of customized host-microbiome co-metabolism.
Introduction
The human microbiome is crucial to health and well-being, and alterations in the gut and skin microbiomes have been connected to a number of illnesses. Genetics, sex, diet, lifestyle, geography, and ethnicity are some of the variables that affect the composition of microbiomes, which differ by body site and individual. A Westernized lifestyle has been linked to negative changes in the makeup of the microbiome, including the loss of beneficial taxa. This lifestyle is typified by an industrialized environment, a diet high in sugar, a lack of exercise, poor sanitation, and excessive use of antibiotics. Conducting research on the microbiomes of non-industrialized communities may aid in the analysis of how the Westernized lifestyle affects the microbiome. Nevertheless, little is known about people from the global south, which restricts our understanding of their unique involvement in health and illness conditions.
Introducing APOLLO
In this article, researchers introduce APOLLO, a resource of 247,092 microbial genome-scale reconstructions that is as sophisticated as the underlying MAG resources. When combined, they comprise 595 recognized species; the remaining inventory is made up of taxa that are not yet cultivated or investigated. When it comes to the usual taxonomic distribution of the human microbiome, APOLLO performs significantly better than AGORA2.
Researchers have to greatly optimize and parallelize the reconstruction process in order to provide this extensive reconstruction resource. Likewise, computing difficulties have to be resolved in order to allow the analysis and simulations using the full reconstruction resource. Researchers used high-performance computers and crowdsourcing to do this. Even though this dataset is currently incomplete since new microorganisms are continuously being discovered by more recent MAG research, researchers show that metabolic reconstruction and modeling techniques can keep up with the rate of genome discovery.
MAG resources, particularly strains from Asian populations that are currently underrepresented in APOLLO, can be comprehensively reanalyzed using a computationally efficient workflow. The pipeline can be used for microbiomes that are not human, like those of wild animals and various Earthly habitats.
Through genome reconstruction and metabolic modeling, the pipeline can clarify the composition and operation of microbial ecosystems that are mainly uncultivated. But depending on how many MAGs are rebuilt, the pipeline takes weeks to months to complete and is computationally and temporally heavy. A single integrated computational tool that allows for draft reconstruction and refinement straight from MAGs could be used in future efforts to optimize the pipeline.
Performance of APOLLO Against Other Previous Genomic Resources
APOLLO clearly outperforms earlier genome-scale reconstruction resources like AGORA2 in terms of displaying the usual taxonomic distribution of the human microbiome. Given that the majority of human microbial taxa are members of the Bacteroidia and Clostridium classes, APOLLO makes it possible to examine the distribution of metabolic features in these significant taxa in a methodical manner.
An additional benefit of APOLLO is that it can identify genomes that are currently unidentified; 7,632 of them are unclassified, even at the phylum level. Verrucomicrobia and other taxa that are currently understudied account for a sizable portion of reconstructed genomes.
There is just one species in this phylum that has been thoroughly investigated before, Akkermansia muciniphila. However, the APOLLO collection has 1,282 strains of Verrucomicrobia that are not identified at the species level, offering a unique chance to study them in silico and then in vitro.
Future Applications of APOLLO
Future research could shed light on the metabolic capacities of the poorly understood and completely unidentified taxa by analyzing the APOLLO reconstructions. Comparing topological characteristics, including metabolite connections and metabolic capacities across species, systematically is another possible use for APOLLO. For example, AGORA has been used in the past to evaluate metabolic heterogeneity among strains and in silico vitamin auxotrophies across taxa using topological aspects of the reconstructions. Such applications will become possible on a far greater scale and scope, thanks to APOLLO.
Conclusion
The metabolic capacities of MAGs have been investigated using a computational pipeline that allows for hitherto unheard-of levels of detail in strain- and molecule-resolved investigation. Using two large-scale MAG resources, the pipeline has produced more than 240,000 genome-scale reconstructions. The classifier, which was created for MAGs from various sources, accurately predicts taxonomical associations. Important responses that categorize taxa are found. Fourteen thousand customized microbiome models were generated and examined to examine the reconstructed strains in their natural habitat. Based on age group, location, body site, and health state, these models could extract functional differences in reaction and route content between people. Further development of the pipeline could eventually enable researchers to fully utilize the growing amount of MAGs and metagenomes that are made publically available and offer fresh perspectives on microbial habitats that are now poorly understood.
Article Source: Reference Paper | Reference Article.
Disclaimer:
The research discussed in this article was conducted and published by the authors of the referenced paper. CBIRT has no involvement in the research itself. This article is intended solely to raise awareness about recent developments and does not claim authorship or endorsement of the research.
Follow Us!
Learn More:




Deotima is a consulting scientific content writing intern at CBIRT. Currently she's pursuing Master's in Bioinformatics at Maulana Abul Kalam Azad University of Technology. As an emerging scientific writer, she is eager to apply her expertise in making intricate scientific concepts comprehensible to individuals from diverse backgrounds. Deotima harbors a particular passion for Structural Bioinformatics and Molecular Dynamics.











{kind=link}