
Small molecule drug discovery could be revolutionized by molecular docking tools. Still, current molecular docking methods are slow to screen targets, leading to missed candidates and unexpected side effects in clinical trials, despite the potential benefits of this approach. Researchers from the University of Warsaw and others present RAPIDDOCK, an effective transformer-based model for blind molecule docking, to close this gap. With a 100x speed advantage, the molecular docking technique RAPIDDOCK beats current approaches on DockGen benchmarks. It has an average inference time of 0.04 seconds on a single GPU and reaches 52.1% and 44.0% success rates on the Posebusters and DockGen benchmarks, respectively. A personalized loss function, pre-training on protein folding, and relative distance embeddings of 3D structures are important characteristics.
Introduction
Medicine will be transformed by speeding up the drug discovery process. Since the majority of new medications are small compounds, a number of deep-learning techniques have been put forth to make the process of docking these molecules to druggable protein targets more efficient. None of these techniques are quick and accurate, despite their impressiveness. In reality, a chemical must be screened against thousands of proteins to get a complete picture of its effects. On the other hand, cutting-edge techniques report run times on the order of seconds per protein on a single GPU. It would therefore take years, even with hundreds of GPUs, to scan a tiny database of one million compounds against the proteome, or all of the proteins in the human body. Such timelines are unacceptable in the process of developing new drugs.
Understanding RAPIDDOCK
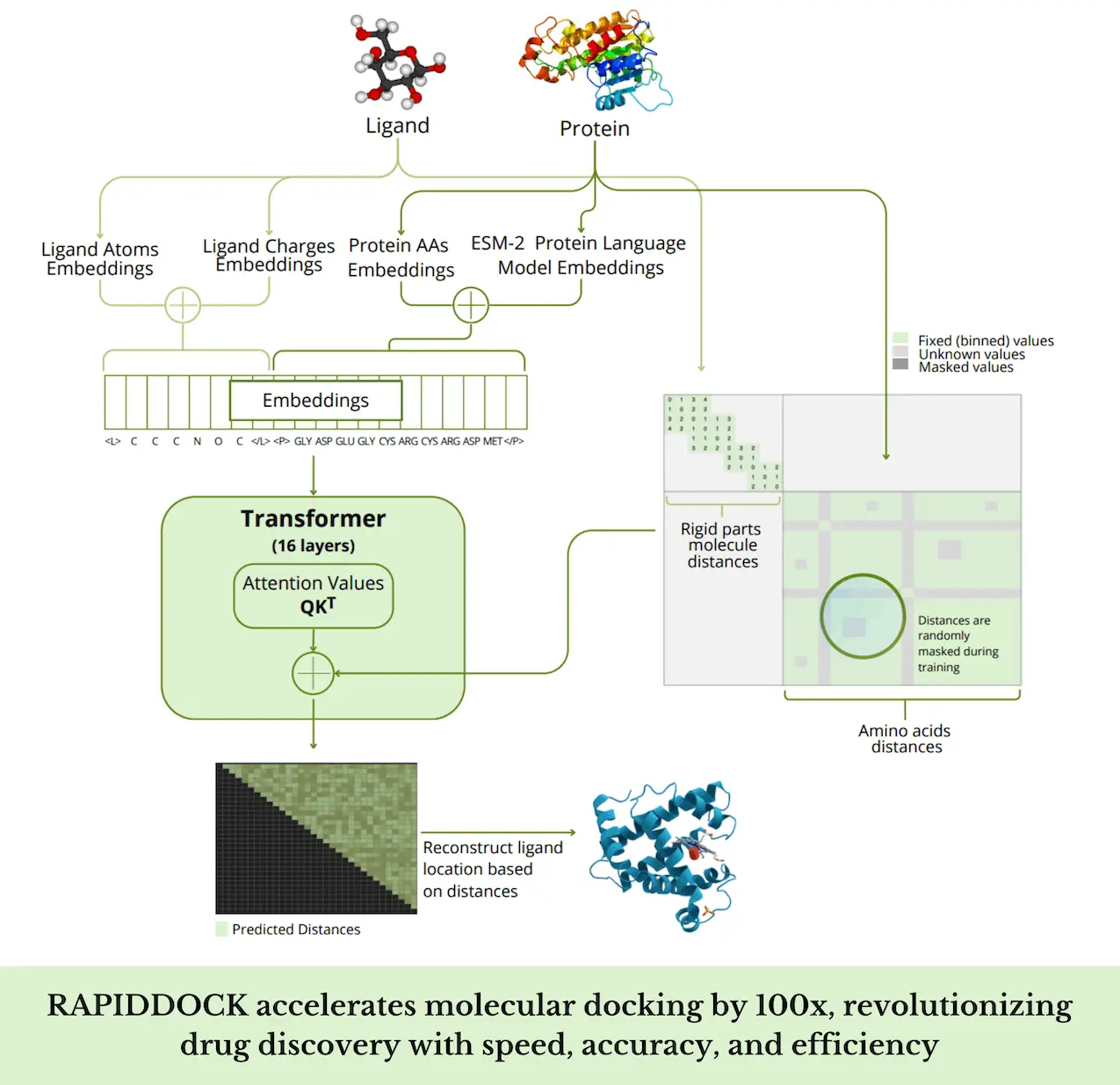
RAPIDDOCK is a transformer-based model that uses a single GPU to complete molecular docking in a single forward pass in a matter of a hundredth of a second. To apply RAPIDDOCK to uncharted protein targets, it uses unbound, perhaps computationally folded proteins to attempt blind docking. This method predicts the final protein-molecule complex’s pairwise distances, including the atom-atom and atom-amino acid distances, given the three-dimensional (3D) structures of the molecule and the protein.
The average runtimes of RAPIDDOCK are 0.03 and 0.05 seconds, respectively, and it achieves success rates (i.e., RMSD < 2Å) of 52.1% on the Posebusters benchmark and 44.0% on the DockGen benchmark. Compared to the latest DiffDock-L (22.6% success rate, 35.4 seconds on DockGen) and the NeuralPLexer (22.6% success rate, 3.77 seconds on Posebusters), the approach performs noticeably better. RAPIDDOCK can facilitate new research directions and use cases due to its speed and precision. Docking ten million molecules to all human proteins on a cluster with 512 GPUs would take nine days with RAPIDDOCK, whereas DiffDock-L would take roughly 20 years, or perhaps 200 years, with a computationally demanding approach like AlphaFold-3.
Key Features of the Study
- Researchers present RAPIDDOCK, a transformer-based model that is the first of its kind and can accurately perform high-throughput molecular docking.
- Researchers use the most difficult benchmarks to test RAPIDDOCK. With RMSD < 2Å on Posebusters and 44.0% on DockGen, the model docks 52.1% of samples, making it one of the most accurate techniques. Crucially, RAPIDDOCK is at least 100× faster than similar techniques by requiring an average of only 0.04 seconds for inference on a single GPU.
- The researchers outline the design decisions contributing to RAPIDDOCK’s success and offer ablations.
Training of RADIPDOCK
A protein folding task was used to train the 16-layer, 512-hidden-size RAPIDDOCK model to enhance docking performance. To accurately replicate the protein folding job, the model was trained to estimate amino acid distances with a 97% masking factor. It took almost three days on a computer with eight A100 GPUs to refine the model on the molecular docking job on the training dataset. Models with the same number of parameters that were shallower but wider did not perform as well as RAPIDDOCK’s deep and narrow architecture.
Future Work
The future plan is to provide a confidence score for RAPIDDOCK’s predictions, which is essential for making decisions. Another external light model can be trained to accomplish this, but ideally, RAPIDDOCK will provide its own confidence score. Additionally, the strategy emphasizes identifying non-binding ligands, optimizing RAPIDDOCK for ligand-protein binding strength prediction, and effectively detecting potentially harmful drug interactions throughout the human proteome. Training larger models on larger datasets and carrying out in-depth research on scaling qualities are also part of the agenda. The PLINDER protein-ligand datasets will be used for primary training, and pre-training will be expanded to more than 200 million protein structures predicted by AlphaFold 2.
Conclusion
A molecular docking model that is incredibly quick and precise, RAPIDDOCK creates new opportunities for in silico drug creation. The model is well-versed in the physicochemical principles underlying the formation of biological structures, having been trained in both protein folding and molecular docking. It achieves precise results in molecular docking on both holo- and apostructures. RAPIDDOCK is expected to give drug makers a thorough understanding of possible drug interactions throughout the human proteome in the near future. This will open up significant research directions in machine learning and biology. Last but not least, the transformer-based design makes the model ideal for extending pre-training to related biological tasks or refining downstream tasks like cell-level drug interaction or toxicity prediction, which is one of the current research interests.
Article Source: Reference Paper
Important Note: arXiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Follow Us!
Learn More:




Deotima is a consulting scientific content writing intern at CBIRT. Currently she's pursuing Master's in Bioinformatics at Maulana Abul Kalam Azad University of Technology. As an emerging scientific writer, she is eager to apply her expertise in making intricate scientific concepts comprehensible to individuals from diverse backgrounds. Deotima harbors a particular passion for Structural Bioinformatics and Molecular Dynamics.











{kind=link}