
Single-molecule Next-Generation Protein Sequencing (NGPS) provides a powerful alternative to mass spectrometry for analyzing protein variants, allowing detailed identification of proteoforms and amino acid changes at the individual molecule level. New data analysis techniques and bioinformatic tools must be developed to handle the unusual data type produced by NGPS. Here, scientists from Quantum-Si introduce ProteoVue™, a complete bioinformatics pipeline that uses the Quantum-Si Platinum® NGPS technology for Single Amino Acid Variant (SAAV) identification and quantification. The system known as ProteoVue combines several analytical elements, such as identification segment detection, fluorescent dye categorization, pulse-calling, and a library of kinetic signatures powered by neural networks. In fundamental research, biomarker identification, and clinical applications, ProteoVue offers a basis for sophisticated variant analysis as NGPS technology advances.
Introduction
A notable development in proteomics is Quantum-Si’s Next Generation Protein Sequencing (NGPS) technology, which is accessible on the Platinum platform. Individual peptides, including isobaric amino acids and highly identical proteoforms, can be measured in real-time using NGPS, as opposed to mass spectrometry (MS). This orthogonal approach allows the analysis of proteoforms, which are protein variants resulting from genomic, transcriptomic, and post-translational variation, and complements numerous well-established proteomics techniques. NGPS creates new opportunities for studying proteoforms by directly analyzing individual protein molecules using single-molecule measurements, offering comprehensive details on their variations and changes. The comprehension of proteoforms and their function in biological and disease pathways is improved by this orthogonal tool, which supports numerous well-established proteomics techniques.
Understanding ProteoVue
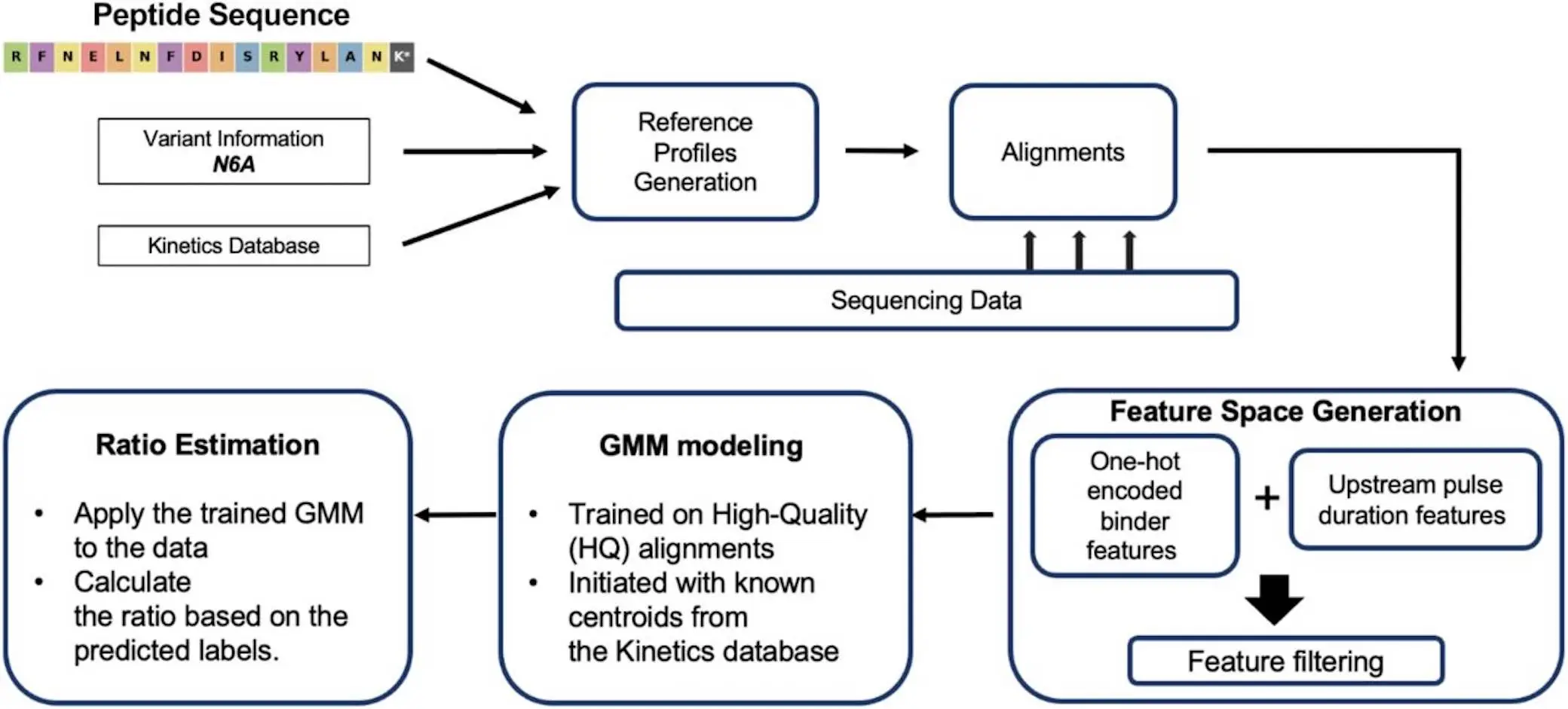
ProteoVue is a machine-learning technology that creates reference profiles using peptide variations as inputs. Clusters of pulse durations are segmented and assigned to variants after it extracts the anticipated peptide states and pulse duration profiles from the kinetics database. After aligning sequential data from a binary mixture run to the reference for every variant, positional kinetics are aggregated, and kinetic characteristics such as PD are extracted.
To capture recognizer read variance and the complete pentameric context impacting pulse duration, ProteoVue integrates aggregated positional kinetics with one-hot encoded binary recognizer characteristics to create a multidimensional feature space. These attributes are used to train a two-component GMM, with the expected kinetic profiles and recognizer identities serving as guidance for the initial centroids. Applying the trained GMM to the entire dataset yields predicted population identities for all data points. ProteoVue calculates the ratio of variant populations from the GMM populations.
By precisely identifying many variants based on minute variations in single-residue substitutions, the ProteoVue pipeline efficiently recovers variant peptide populations in regulated mixes. The pipeline is useful for extracting information from sparse data because its kinetic description of recognition segmentation can accommodate variants with different kinetic and recognizer-binding qualities. A neural network-based kinetic database is integrated into the pipeline to further refine the scoring and allow it to predict pulse duration distributions for pentameric sequence motifs that have not yet been observed. The pipeline’s strength is its ability to handle variants with various kinetic and recognizer-binding properties.
Researchers show that ProteoVue can recover anticipated variant ratios for various substitution types, including residues without direct amino acid recognizers. The pipeline continuously captures the essential kinetic characteristics needed for variant discrimination despite the fact that some extreme examples are still difficult to handle. This highlights the pipeline’s promise as a flexible and potent tool for proteomic research. ProteoVue offers the basis for more sophisticated variant analysis in fundamental research, biomarker identification, and clinical applications as NGPS technology advances and recognizer libraries grow.
Limitations
A technique known as recognizer-based clustering makes use of a recognizer to find and categorize variations according to their recognition. Its discriminative power may be weakened by issues like combining variants with the same recognizer. Furthermore, it has trouble telling entirely invisible variations apart, particularly in situations with extreme ratios. These difficulties point to areas in need of development, including kinetic model refinement, recognizer library expansion, and the use of complementary biochemical techniques.
In the future, ProteoVue’s development will concentrate on improving the accuracy and richness of the kinetic characteristics. Rare variants or variants in complicated proteomes are examples of challenging cases that may be better resolved with more experimental data and better modeling. Variant discrimination under challenging situations may be enhanced by combining more complex statistical frameworks, different machine learning techniques, and adaptive thresholding. Furthermore, one of the most critical steps in assessing ProteoVue’s efficacy in the context of high sample complexity will be to apply it to biological samples rather than synthetic peptides.
Conclusion
Using NGPS, the bioinformatics pipeline ProteoVue was created to identify and measure peptide variations. This technique measures SAAVs and other protein variants at the amino acid level using single-molecule sequencing. To overcome the difficulties associated with analyzing data from single-molecule protein sequencing, ProteoVue combines clustering-based variant calling, alignment methods, and sophisticated signal processing. This pipeline shows that reliable variant calling in single-molecule protein sequencing is feasible. As NGPS develops, ProteoVue bioinformatic techniques will continue to be developed, allowing it to reach its full proteomics potential.
FAQ
Next-Generation Protein Sequencing (NGPS) is an innovative technology that analyzes proteins at the single-molecule level. Unlike traditional mass spectrometry methods, NGPS can directly examine individual protein molecules to identify specific variations in their structure, including different proteoforms (variations of the same protein) and amino acid substitutions. This high-resolution approach provides researchers with unprecedented detail about protein composition and modifications, making it particularly valuable for understanding protein diversity in biological systems. The technology’s ability to detect subtle molecular changes offers significant advantages in fields such as drug development, disease research, and protein engineering, where precise understanding of protein structure and variation is crucial.
Article Source: Reference Paper
Disclaimer:
The research discussed in this article was conducted and published by the authors of the referenced paper. CBIRT has no involvement in the research itself. This article is intended solely to raise awareness about recent developments and does not claim authorship or endorsement of the research.
Important Note: bioRxiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Follow Us!
Learn More:




Deotima is a consulting scientific content writing intern at CBIRT. Currently she's pursuing Master's in Bioinformatics at Maulana Abul Kalam Azad University of Technology. As an emerging scientific writer, she is eager to apply her expertise in making intricate scientific concepts comprehensible to individuals from diverse backgrounds. Deotima harbors a particular passion for Structural Bioinformatics and Molecular Dynamics.











{kind=link}