
Our cells are supported critically by proteins, which are also highly affected by their 3D structure. The triumph in computational biology is the ability to predict these structures from a chain of amino acids belonging to protein. Nevertheless, a protein is not static as it can take different shapes, like a dancing girl. Understanding this collection of shapes called conformations well is crucial for designing new medicines and understanding how proteins work. This is where Fan et al.’s approach, Diffold, comes in. This is because it utilizes diffusion models to study the landscape constituted by any protein configuration.
Diffold: A Diffusion Odyssey through Protein Conformational Space
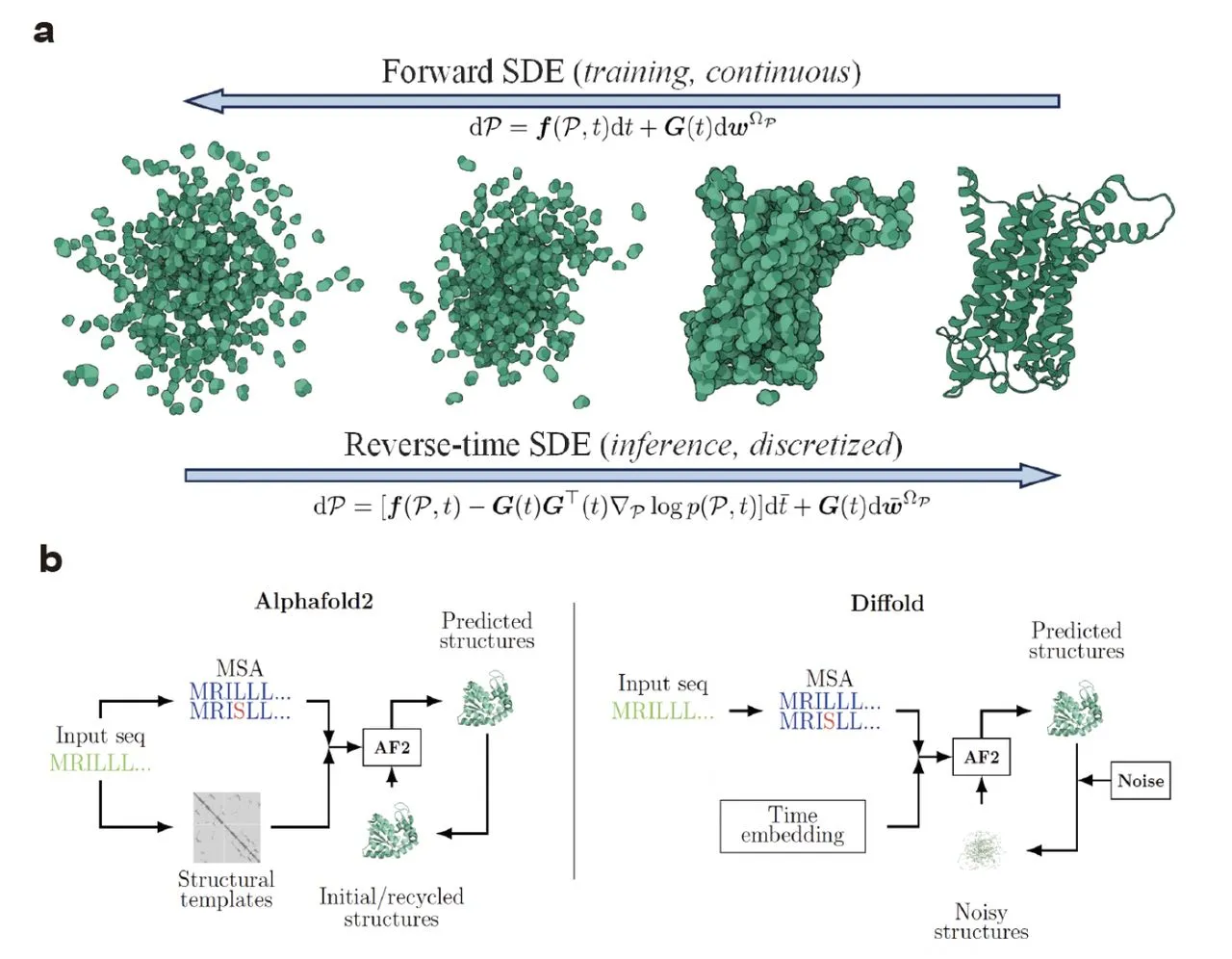
Building on the success of AlphaFold2, Diffold also utilizes diffusion models, a machine-learning technique that allows exploration into various forms of protein conformation. Consider a protein’s structure as a huge landscape. Thus, diffusion models enable Diffold to take a random walk over this terrain, thereby gradually discovering many different conformations with different probabilities.
Here is a deeper look into how Diffold works:
AlphaFold2 as a Basis: The fundamental architecture of AlphaFold2, which takes protein sequences as inputs and outputs structure predictions, is used by Diffold. However, in this case, the same has been modified to handle the diffusion process.
Backbone Modelling: The main structure of the protein – the backbone – is broken down by Diffold into its component positions and rotations. These components allow for independent diffusion that can better explore conformational space.
Reweighting the Landscape: Certain biases come with protein data; some families, for example, get more attention than others. To address this concern, Diffold uses a hierarchical reweighting scheme during training that ensures all protein families equally participate in the diffusion process.
Custom Loss Function: Determining whether or not Diffold has achieved its exploration goals involves developing an appropriate loss function. This takes into account both overall form and relative positioning for translation and rotation of parts constituting proteins, thus offering a more precise measure of the conformation sampled.
Beyond a Static Snapshot: The Power of Diffold
Diffold makes Protein Research Exciting:
Unveiling the Diversity of Conformations: Diffold provides a comprehensive view of protein flexibility through sampling various conformations. It is important to know how proteins interact with other molecules and how mutations affect their functions.
Protein Dynamics Simulation: Langevin dynamics is a method for simulating the motion of a protein molecule over some time. Based on its connection with diffusion models, Diffold offers an opportunity for Langevin dynamics simulations. This enables studying larger-scale protein dynamics.
Bridging the Conformational Gap: Researchers can visualize continuous transitions between two known protein conformations through structural interpolation. Deep insights into how proteins change their shape are permitted by this process, which is enhanced by the Diffold system.
Diffold Indistinguishable: A Case Study
The researchers demonstrate the effectiveness of Diffold in a real-world setting. The protein SLC15A4 has an important role in cellular activities and exists in two distinct states. So far, one could not determine which of these forms was essential for binding feeblin, a specific inhibitor molecule. On the contrary, Diffold succeeded in sampling both forms of SLC15A4 and was able to show which one interacts with the feeblin. In drug discovery research, this proves that finding the right protein conformation is necessary for effective drug design.
Future Perspectives on Protein Conformation Sampling
Diffold brought about considerable improvements in protein conformation sampling. As time goes on, further advancements can be expected:
Beyond Backbones: The protein backbone is the main interest in Diffold, and considering the future releases of the same software to factor in side chain fluctuations more explicitly would yield a complete picture of protein flexibility.
Multimeric Marvels: Most often, proteins act together through the formation of complexes. Consequently, by including protein-protein interactions into Diffold, it may be possible to explore these complex supramolecular entities further.
Merging with Sequence Magic: Diffusion-based methodologies such as Diffold combined with various sequence manipulation techniques can help explain how changes in primary structure result in conformational alterations.
Join the Conversation!
Diffold marks the beginning of a deep examination of the problem concerning protein functions and dynamic behaviors at larger molecular scales. This blog has only skimmed the surface.
What do you think about Diffold and its prospective effects on bioinformatics?
Please share your comments and questions below – let us keep the conversation going.
Article Source: Reference Paper | Diffold’s code and service will be made available by the authors upon publication.
Important Note: bioRxiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Follow Us!
Learn More:




Anchal is a consulting scientific writing intern at CBIRT with a passion for bioinformatics and its miracles. She is pursuing an MTech in Bioinformatics from Delhi Technological University, Delhi. Through engaging prose, she invites readers to explore the captivating world of bioinformatics, showcasing its groundbreaking contributions to understanding the mysteries of life. Besides science, she enjoys reading and painting.











{kind=link}