
In a breakthrough that could transform drug discovery, a team of researchers from ShanghaiTech University has introduced PhysDock, a novel AI-powered model designed to predict how small molecules (ligands) bind to proteins with atomic-level precision.
Protein-ligand docking plays a central role in structure-based drug design. It helps researchers understand how drugs interact with their biological targets, paving the way for developing more effective and selective therapies. However, existing computational docking tools often trade physical accuracy for speed, or vice versa. PhysDock changes that by marrying deep learning with molecular physics, offering a robust and efficient solution to this long-standing problem.
What is PhysDock?
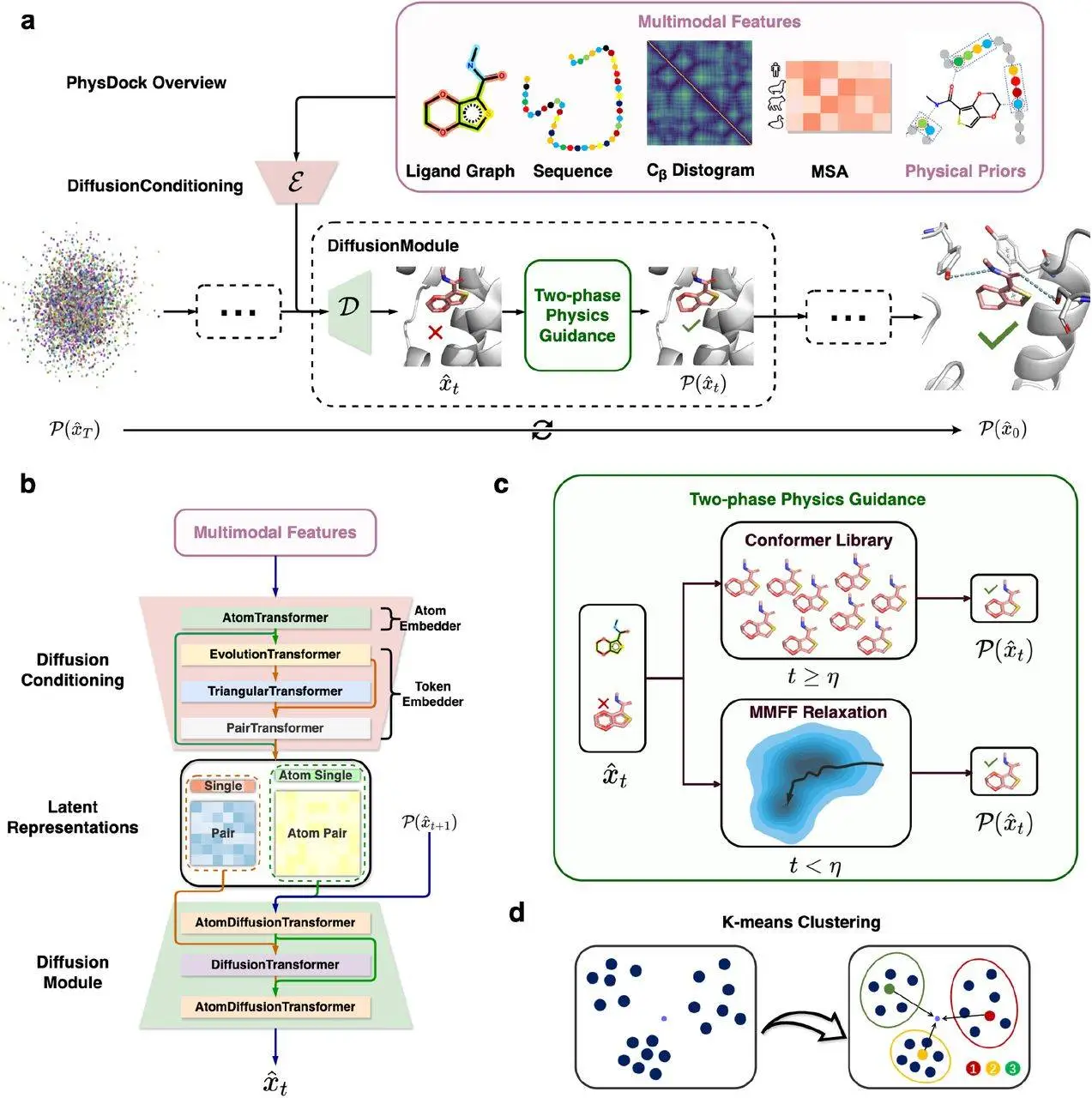
Diffusion of ligand poses refinement via physics constraints, such as molecular force fields and conformer libraries, is also integrated, ensuring compliance with the look and follow chemical rules paradigm. With PhysDock, a physics-informed, all-atom diffusion model, the three-dimensional structures of protein-ligand complexes are predicted. Generatively creating models and employing machine learning principles with deepfakes of the assumed likeness yields agile, flexible-skeletal molecular predictions.
How PhysDock Works?
The PhysDock pipeline operates in two stages. In the first, it generates potential ligand conformations using an all-atom diffusion model conditioned on protein structure and interaction information. In the second stage, a two-phase physical guidance mechanism is applied to refine the predicted structures, ensuring correct chirality, proper bond lengths, and the removal of steric clashes. This process is iterative and incorporates multimodal features, such as molecular graphs, protein sequences, and distance-based maps, to improve precision. The model also uses clustering algorithms to identify the most representative and energetically favorable structures from the generated samples.
What Makes PhysDock Unique
What separates PhysDock from the rest is the incorporation of both the ligand flexibility and the protein “precision-flexibility,” which is the subtle side-chain movements proteins have while undergoing ligand binding. The majority of other models work on the problem with the protein kept rigid, which reduces accuracy. PhysDock is also unique because it doesn’t use only data in the learning process, but instead combines and relies on certain known interaction types like hydrogen bonds and hydrophobic contacts. Besides those, it also uses SE(3)-diffusion modeling, which guarantees invariance of prediction within spatial transformations and is vital in 3D space modeling.
PhysDock was evaluated on gold-standard datasets including the codenamed PoseBusters, DeepDockingDare (DDD), and the newly compiled PhiBench. It outperformed classical docking methods such as Glide and AutoDock Vina and modern deep learning methods, including DiffDock and AlphaFold3, particularly in redocking and cross-docking scenarios. In redocking, it achieved high accuracy in recovering known ligand poses, while in cross-docking, where ligands are docked into different conformations of the same protein, it demonstrated superior generalization, maintaining strong performance even without prior binding pocket information.
Real-World Applications
PhysDock has demonstrated its capabilities in practical scenarios. The team employed it to study the binding selectivity of CB1 and CB2 cannabinoid receptors, two similar yet different proteins that serve distinct functions in the human body. PhysDock was able to predict their ligand binding and selectivity’s discriminative traits’ molecular structures. In another instance, it was used for virtual screening of over 335,000 compounds for NTRK3 kinase, a cancer-associated protein. PhysDock was able to differentiate known drug candidates from weak binders and even proposed new high-affinity structural propositions with explanatory details.
Conclusion
PhysDock illustrates the stunning combination of physics with AI in a new paradigm of estimating protein-ligand complexes. It surpasses best practices in accuracy, efficiency, and physical realism in computation. Moreover, its use in receptor selectivity and virtual screening showcases its practicality, while its design paves the way for later large-scale integrations into drug pantries. The enhancement of molecular modeling tools has brought forth expectations for efficiency, PhysDock is advancing research in bioinformatics and pharmaceuticals.
Article Source: Reference Paper
Disclaimer:
The research discussed in this article was conducted and published by the authors of the referenced paper. CBIRT has no involvement in the research itself. This article is intended solely to raise awareness about recent developments and does not claim authorship or endorsement of the research.
Important Note: bioRxiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Follow Us!
Learn More:




Anchal is a consulting scientific writing intern at CBIRT with a passion for bioinformatics and its miracles. She is pursuing an MTech in Bioinformatics from Delhi Technological University, Delhi. Through engaging prose, she invites readers to explore the captivating world of bioinformatics, showcasing its groundbreaking contributions to understanding the mysteries of life. Besides science, she enjoys reading and painting.











{kind=link}