
Virtual screening techniques, which concentrate on binding affinities, are becoming more and more popular for early-stage drug development. Although deep learning-based methods predict binding affinities, limited training data raises questions about their generalization capabilities in large chemical spaces. In this study, scientists from the Department of Chemistry, KAIST, Republic of Korea, provide PharmacoNet, the first deep-learning framework for modeling pharmacophores to achieve incredibly quick virtual screening. The efficacy of ligands was assessed in a study using PharmacoNet, a protein-based pharmacophore model. High generality between targets and ligands is guaranteed by the model’s parameterized analytical scoring function. By discovering selective inhibitors from 187 million chemicals against cannabinoid receptors in 21 hours on a single CPU, the study showcased the speed and precision of PharmacoNet and the potential of deep learning in pharmacophore modeling.
Introduction
The exploration of large chemical regions is required to improve the hit rate of drug discovery. The number of libraries has increased from millions to billions, making molecular docking an essential evaluation method. However, because molecular docking requires a high computational cost and takes seconds to minutes, effectively screening huge volumes is difficult.
Pre-screening techniques are crucial for finding inhibitors in a library that show promise. Effective molecular exploration is the primary goal of these techniques, which include Bayesian searches and structured library searches. These searches aid in finding inhibitors in sparse chemical spaces. However, these tactics may miss good hits, which emphasizes the need for a strategy that strikes a balance between speed and accuracy. It is possible to find difficult inhibitors by using a tiered method that evaluates the entire library. Ultimately, effective pre-screening techniques and chemical exploration are essential for the successful discovery of inhibitors.
PharmacoNet: An Overview
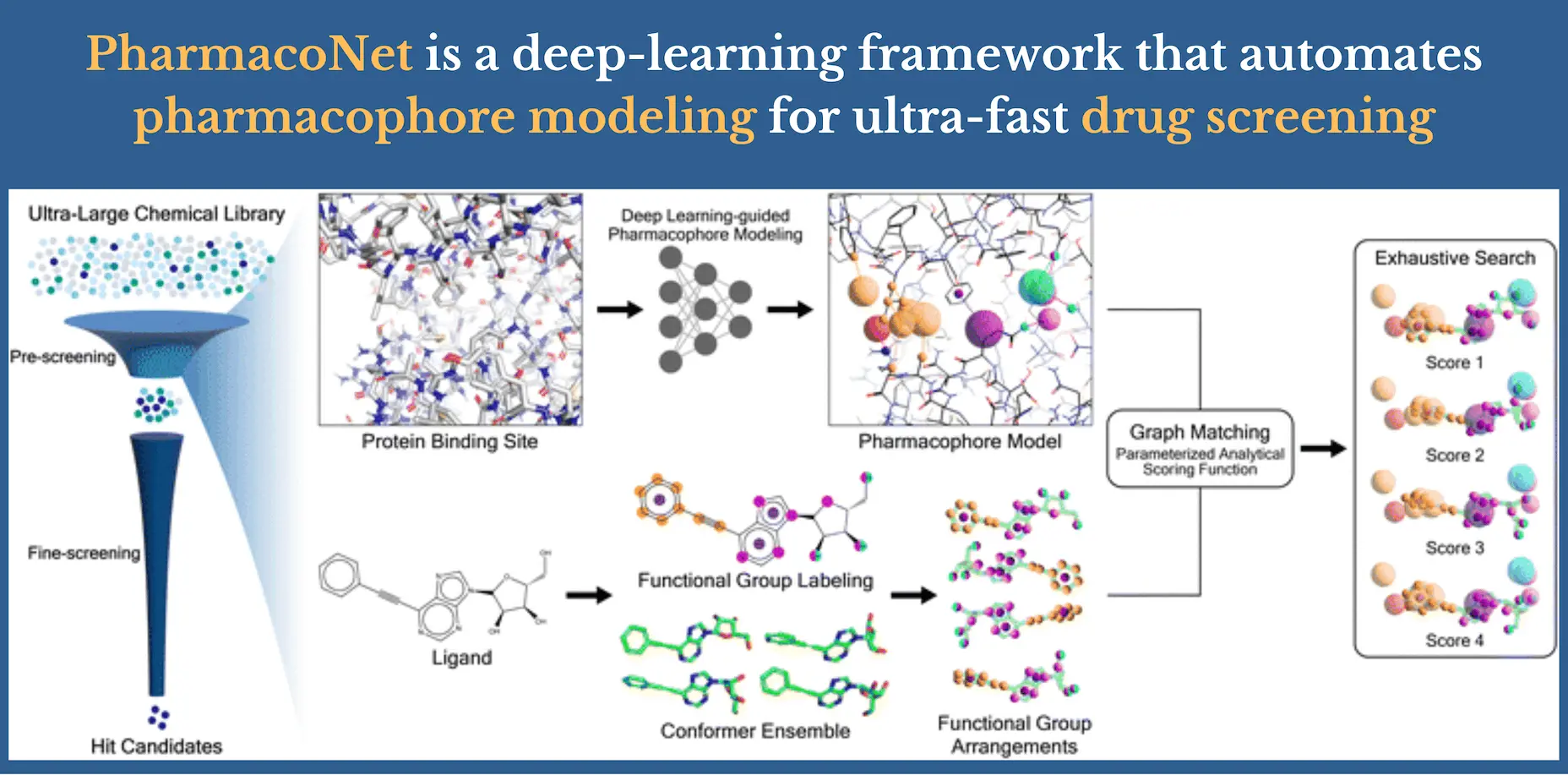
The first deep-learning framework for modeling protein-based pharmacophores is called PharmacoNet. In order to automatically identify important protein functional groups (hotspots) and the best places for matching pharmacophore points in order to build a pharmacophore model, PharmacoNet presents instance segmentation DL modeling. PharmacoNet then uses an algorithm to determine the ligand’s compatibility with the pharmacophore at the NCI level, incorporating a parameterized analytic function. By focusing on pharmacophoric interaction instead of atomistic interaction, the method significantly lowers processing needs while maintaining a respectable level of accuracy. Additionally, by avoiding the over-fitting that occurs when deep learning models have too many parameters, this coarse-grained evaluation technique guarantees its dependability and applicability across a variety of chemical spaces.
PharmacoNet is incredibly quick and somewhat accurate; in virtual screening benchmarks, it achieved 3,000-fold speedups while still performing competitively when compared to traditional docking techniques like AutoDock Vina. Additionally, PharmacoNet used a desktop computer with a single 32-core CPU to analyze 187 million compounds in order to find possible Cannabinoid (CB) antagonist candidates with potency and CB2/CB1 selectivity in 21 hours (11 years for AutoDock Vina). By facilitating the quick screening of large chemical libraries, the findings show that PharmacoNet can expedite drug discovery. OpenPharmaco, a comprehensive graphical user interface (GUI) program offered by PharmacoNet, is also intended to make it easier for a wide range of users—including those lacking computational resources and experience—to use protein-based pharmacophore modeling and high-throughput virtual screening.
PharmacoNet Framework
Three steps make up PharmacoNet: 1) DL-based pharmacophore modeling, 2) coarse-grained graph matching, and 3) rating based on distance likelihood. The pharmacophore model is first built by PharmacoNet, utilizing only the structural details of a target protein binding site. It does this by identifying the hotspots and the best places (pharmacophore point) for ligand functional groups to form stable NCIs with each hotspot. Next, the spatial link between ligands and the pharmacophore model is efficiently estimated by the graph-matching technique. In comparison to the analogous atomistic prediction, this pharmacophore-level prediction necessitates substantially less computation. Finally, the pharmacophore-level abstraction of PLIs allows the scoring function to provide the binding affinity of each pose with a high degree of generalization and a respectable degree of accuracy.
Limitations
Even with these developments, there is still room for improvement. PharmacoNet makes quick and accurate predictions using a scoring and graph-matching method at the pharmacophore level. Nevertheless, there is a cost associated with this pharmacophore-level abstraction. For instance, it is unable to distinguish between variations in the strength of the same salt bridge, which are ascribed to minute variations in charge and atom type, because it does not capture atom-level properties. Furthermore, it is unable to take intramolecular energy fluctuations into consideration. In this sense, the current method works better for pre-screening and, more precisely, for post-scoring. Atomistic elements should be added to the scoring function while preserving a high degree of generalization ability in order to get beyond these restrictions.
Conclusion
PharmacoNet is a deep learning system that tackles major issues in large-scale virtual screening by modeling pharmacophores. It is a data-driven approach to automate pharmacophore modeling using protein structures through experimental binding structures. Using an algorithmic scoring function, PharmacoNet isolates protein-ligand interactions at the pharmacophore level, enabling quick and accurate scoring and increasing computing speed. The ability to find strong and specific cannabinoid receptor antagonists in 21 hours on a single CPU demonstrates how this enables ultra-largescale virtual screening at reasonable costs. For large-scale virtual screening on desktop PCs, PharmacoNet also offers an intuitive graphical user interface. PharmacoNet presents a fresh approach to deep learning methods for quick and accurate ultra-large-scale virtual screening in drug development. Researchers think this method will make it easier to implement ultra-large-scale virtual screening in practical settings.
Article Source: Reference Paper | The source code, trained models, and GUI software are available on GitHub.
Disclaimer:
The research discussed in this article was conducted and published by the authors of the referenced paper. CBIRT has no involvement in the research itself. This article is intended solely to raise awareness about recent developments and does not claim authorship or endorsement of the research.
Follow Us!
Learn More:




Deotima is a consulting scientific content writing intern at CBIRT. Currently she's pursuing Master's in Bioinformatics at Maulana Abul Kalam Azad University of Technology. As an emerging scientific writer, she is eager to apply her expertise in making intricate scientific concepts comprehensible to individuals from diverse backgrounds. Deotima harbors a particular passion for Structural Bioinformatics and Molecular Dynamics.











{kind=link}