
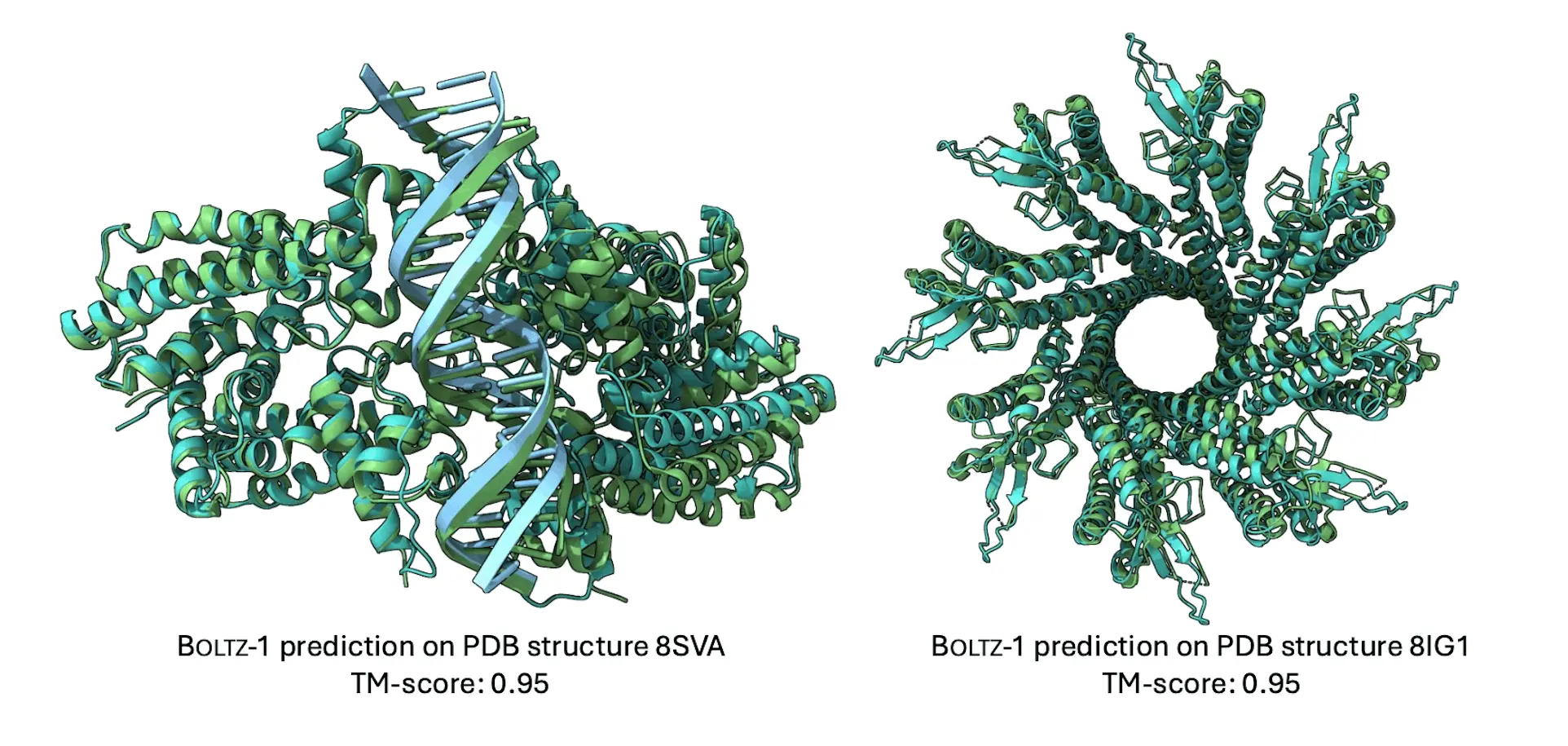
Developing an understanding of biomolecular interactions is essential for developing domains such as protein design and drug development. The open-source deep learning model Boltz-1, which researchers from MIT present in this research, combines advances in model design, speed optimization, and data processing to predict the 3D structures of biomolecular complexes with an accuracy of AlphaFold3. By performing similarly to the most advanced commercial models on various criteria, Boltz-1 establishes a new standard for structural biology tools available for purchase. The goal of making the training and inference code, model weights, datasets, and benchmarks available under the MIT open license is to promote international cooperation, speed up research, and offer a strong foundation for advancing biomolecular modeling.
Introduction
Nearly every biological mechanism is driven by biomolecular interactions, and our comprehension of these interactions informs the creation of novel treatments and the identification of the factors that contribute to disease. In 2020, AlphaFold2 showed that deep learning algorithms can predict single-chain protein structures for a wide class of protein sequences with experimental accuracy. Nonetheless, there was still a crucial query regarding the 3D modeling of biomolecular complexes. The research community has come a long way in the last few years in tackling this important issue. In particular, modeling the interactions between various biomolecules has been accomplished through deep generative models. DiffDock has demonstrated notable advancements over conventional molecular docking techniques, and most recently, AlphaFold3 has achieved previously unheard-of accuracy in predicting arbitrary biomolecular complexes.
Boltz-1: Improving on AlphaFold3’s Foundation
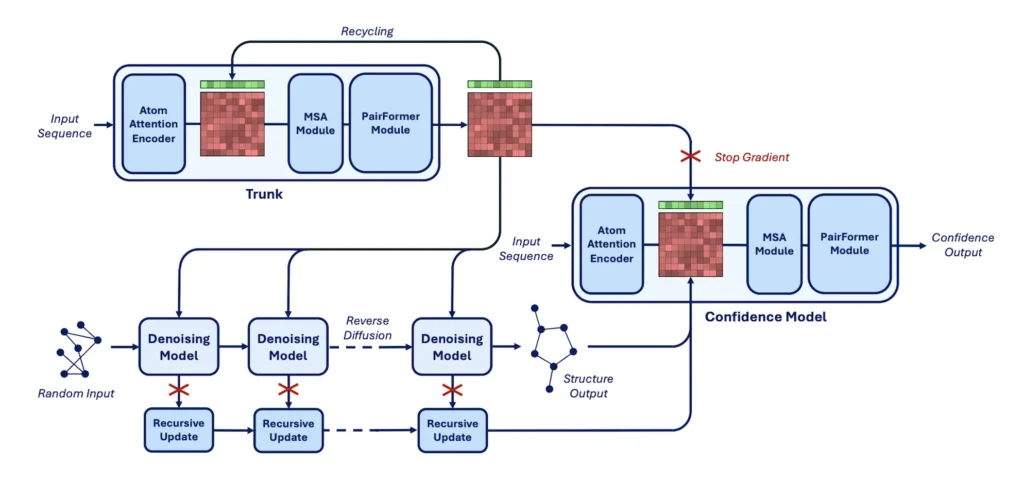
Let’s look into some features of AlphaFold3 as the model training started by reproducing AlphaFold3 architecture. Diffusion model AlphaFold3 denoizes atom coordinates at two levels of resolution: heavy atoms and tokens, using a multi-resolution transformer-based approach. Tokens are described as individual heavy atoms for other compounds and modified residues or bases, nucleic acid bases for RNA and DNA, and amino acids for protein chains. In addition, AlphaFold3 uses a central trunk architecture to establish the attention pair bias of the denoising transformer and initialize the representations of tokens. Although this trunk is designed to be independent of a particular diffusion time or input structure, it is computationally costly because it uses token pairs and axial attention operations.
Understanding Boltz-1
During the Boltz-1 development process, many methods for AlphaFold3’s architecture were tested, and the results showed notable advances. Various stages of the changes were evaluated using a smaller architecture to lower computing expenses. Although direct ablation investigations cannot be carried out, these modifications were anticipated to apply to the final full-sized model.
Boltz-1 is the open-source model that reaches AlphaFold3 accuracy standards, and it is the first model that is completely commercially available; datasets, benchmarks, model weights, and training and inference code are all freely available under the MIT license. New algorithms for effective and reliable MSA pairing, modifications to the architecture’s representation flow, and an update to the confidence model are all included in Boltz-1. This breakthrough enables Boltz-1 experimentation, validation, and innovation by researchers, developers, and organizations worldwide.
Proteins, ligands, and nucleic acids are represented by the Boltz-1 algorithm using genetic sequences, smile strings, and amino acid sequences. It supplements the input with anticipated molecular conformations and multiple sequence alignment (MSA). Due to their limited effect on the performance of big models, Boltz-1 does not contain input templates like AlphaFold3 does. A unified cropping technique that combines contiguous and spatial cropping strategies, a strong pocket-conditioning approach designed for typical use situations, and a new algorithm to couple MSAs for multimeric protein complexes from taxonomy information are all included in the algorithm.
Limitations and Future Directions
Visual examination of Boltz-1 predictions showed that the model’s outputs occasionally displayed hallucinations, with overlapping ligands and chains in the data. Similar ligands with a common substructure and identical polymer chains in big complexes were two common patterns seen in the model’s outputs. The removal of overlapping polymer chains did not completely remove overlapping ligands.
A number of PDB database instances show overlapping ligands within the same structure, which could be different binding molecules or processes. The training data may contain these structures, which could introduce false learning signals. Computational constraints led to the adoption of insufficient training crop sizes, such as 384 and 512 tokens, which may have made it more difficult for the model to capture enough spatial context during training.
There are issues with the model’s structure prediction techniques, especially when it comes to predicting non-physical structures. Using basic heuristics to filter out predictions can help mitigate these problems, but it also adds time to the process. To solve these problems, algorithmic advancements will be included in subsequent generations.
Alternate training or improvement techniques will be investigated to lessen these problems. Making the model and its code publicly accessible will encourage the community to look into other restrictions and suggest creative ways to improve its functionality.
Conclusion
Boltz-1 is the first open-source, commercially available model created by MIT researchers that can accurately predict the three-dimensional structures of biomolecular complexes with an accuracy level comparable to AlphaFold3. Boltz-1 extends and replicates the AlphaFold3 technical report by integrating advancements in architecture, data curation, training, and inference procedures. Empirical validation of the model against AlphaFold3 and Chai-1 has shown that it performs comparably on various test sets, including the CASP15 benchmark. Advanced biomolecular modeling tools are now more widely accessible thanks to Boltz-1’s open-source release, which enables organizations and researchers to use the model for experimentation and innovation.
Article Source: Reference Paper | Reference Article | Code: GitHub.
Disclaimer:
The research discussed in this article was conducted and published by the authors of the referenced paper. CBIRT has no involvement in the research itself. This article is intended solely to raise awareness about recent developments and does not claim authorship or endorsement of the research.
Important Note: bioRxiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Follow Us!
Learn More:




Deotima is a consulting scientific content writing intern at CBIRT. Currently she's pursuing Master's in Bioinformatics at Maulana Abul Kalam Azad University of Technology. As an emerging scientific writer, she is eager to apply her expertise in making intricate scientific concepts comprehensible to individuals from diverse backgrounds. Deotima harbors a particular passion for Structural Bioinformatics and Molecular Dynamics.











{kind=link}