
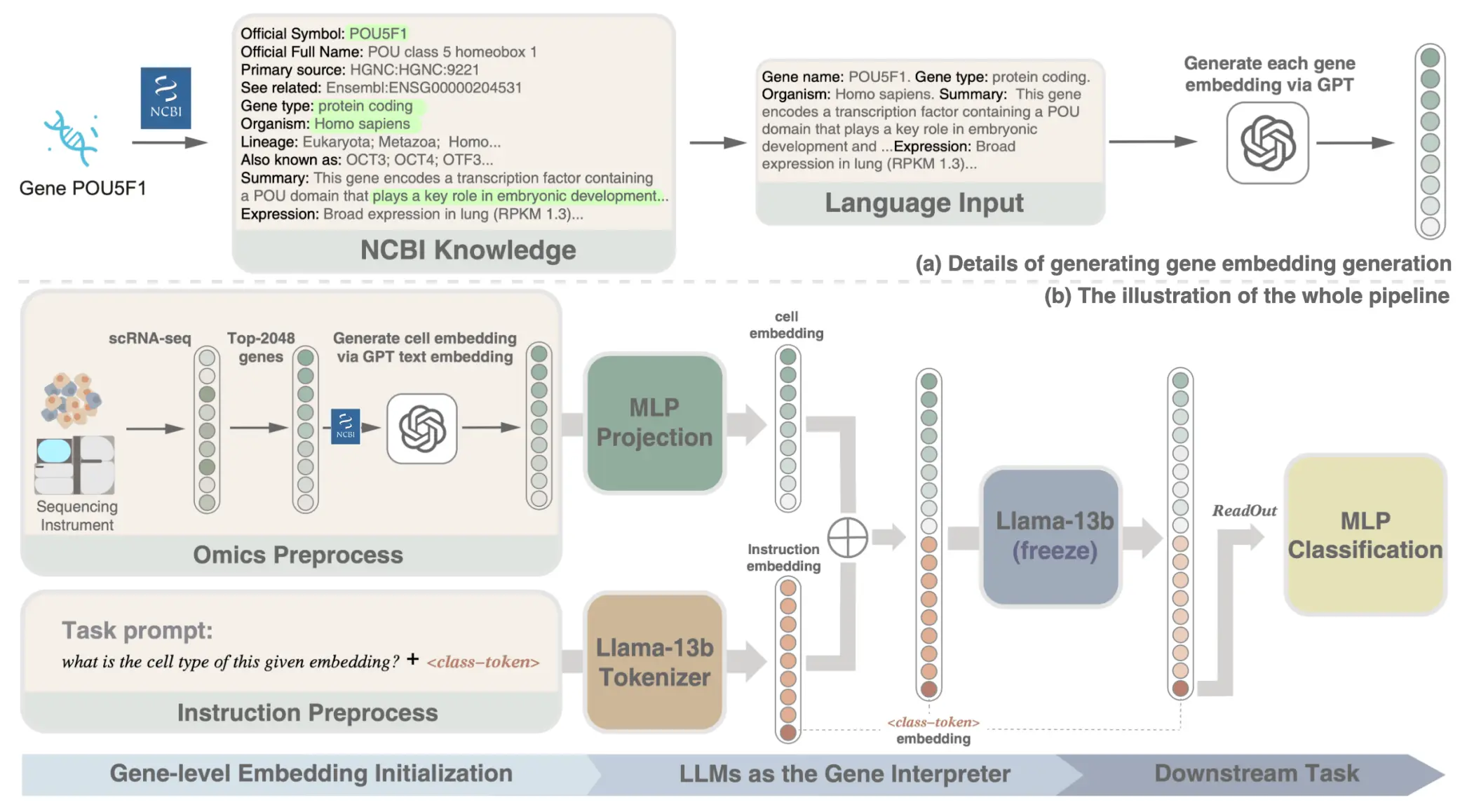
In a groundbreaking study by researchers at the Harbin Institute of Technology, a novel approach to interpreting single-cell RNA sequencing (scRNA-seq) data has emerged. The team, led by experts in computational biology and artificial intelligence, introduced SCREADER—a method that harnesses the power of large language models (LLMs) to transform our understanding of cellular complexity. This work represents a significant leap in bioinformatics, bridging the gap between advanced AI and single-cell biology.
The Need for Better scRNA-seq Interpretation
scRNA-seq technology has opened new frontiers to research in the many different cells that constitute living organisms. However, the enormous and complex multidimensional data produced by scRNA-seq is still complicated to interpret. Microarray or qPCR-based approaches still lack advanced mathematical models that can understand how multiple genes interact and what is the functional outcome of such interactions. The authors from the Harbin Institute of Technology started to address this problem by applying LLMs in a very context-sensitive and interpretational way.
What is SCREADER?
SCREADER is a novel technique that utilizes large language models within the context of scRNA-seq data analysis workflows. In such a case, it improves how single-cell data is interpreted, especially during cell-type classification. More specifically, in its architecture, SCREADER relies on a new training paradigm called prompt-based training, where it can utilize the large amount of biological data contained in LLMs. Consequently, this provides for a more comprehensive analysis of gene expression patterns.
Key Findings and Performance
The researchers conducted extensive experiments to validate SCREADER’s capabilities. They benchmarked it against GenePT, a previously established method, using datasets HUMAN-10k and MOUSE-13k. Across all parameters, SCREADER achieved better results than GenePT in all entities: accuracy, precision, recall, and F1 score.
The increase was, however, more noticeable in the HUMAN-10k sample than in MOUSE-13k. This difference was explained by the larger share of human genes with annotated entries in NCBI knowledge databases (97.5% for humans and 83.6% for mice, respectively). This emphasizes the benefits of using rich biological knowledge for the interpretation of scRNA-seq data.
Challenges in Rare Cell Type Annotation
SCREADER, however, had problems, especially in dealing with rare cell types. For instance, in the MOUSE-13k, Rostral Neurectoderm cells were often mistaken for Parietal Endoderm cells. The researchers explain this by the dataset not being well-balanced, especially as it regards Parietal Endoderm cells, which were few in number. This demonstrates the importance of class imbalance correction in scRNA-seq data to enhance the performance of the models even further.
Visualizing the Impact
The researchers used UMAP (Uniform Manifold Approximation and Projection) to depict how effective SCREADER is. In the beginning, the separation between clusters of cells was minimal, and different types of cells were greatly interfaced. There were some developments in GenePT, but SCREADER appeared to be clustering quite well. Cells of the said type were coherently packed together while other types occupied their own spaces, indicating the model’s capacity to encode similarities of the same class and differences between classes.
For instance, the SCREADER was able to identify distinctly different groups that were intermixed, such as Nascent Mesoderm, Primitive Streak, and Spinal Cord cells. This enhancement in the clustering was due to the confusion matrix, which was able to note a remarkable drop in the number of misclassifications as compared to GenePT.
A Transformative Step Forward
The efficacy of SCREADER is not only useful for cell-type annotations. The embedding of the LLMs in single-cell omics opens new avenues for multi-omics integration and unique cell type detection, which are arguably important areas surrounding precision medicine and developmental biology. By harnessing LLMs’ general knowledge, SCREADER can provide a more sophisticated, knowledge-based understanding of the cell’s complexity.
The findings augment the functional use of artificial intelligence in single-cell biology. SCREADER demonstrates how advanced AI can fill some voids left by classical bioinformatics methods, providing a remarkable improvement in the resolution of cellular studies. The analytical difficulties these researchers are facing likely have the potential to provide a further understanding of nutrition and medicine.
SCREADER is not merely a tool; rather, it reflects the ability of modern research to integrate computation and biology to decode life.
Article Source: Reference Paper
Disclaimer:
The research discussed in this article was conducted and published by the authors of the referenced paper. CBIRT has no involvement in the research itself. This article is intended solely to raise awareness about recent developments and does not claim authorship or endorsement of the research.
Important Note: bioRxiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Follow Us!
Learn More:




Anchal is a consulting scientific writing intern at CBIRT with a passion for bioinformatics and its miracles. She is pursuing an MTech in Bioinformatics from Delhi Technological University, Delhi. Through engaging prose, she invites readers to explore the captivating world of bioinformatics, showcasing its groundbreaking contributions to understanding the mysteries of life. Besides science, she enjoys reading and painting.











 and AI.){kind=link}