
It is essential to connect sequence variation to phenotypic effects to effectively utilize huge genomic datasets. Here, researchers describe a unique method for characterizing spontaneously evolved variations of a rice immune receptor by integrating protein language modeling with directed evolution. The rice immunological receptor Pik-1 was designed to avoid detection by existing Pik-1 alleles by binding and recognizing the fungal proteins Avr-PikC and Avr-PikF. Sequence variation and ligand binding behavior were correlated using a refined protein language model. In vitro tests revealed that two variants had better ligand binding than the wild-type Pik-1 receptor, as seen by their high binding scores against Avr-PikC. This machine-learning method has the potential to be useful for investigating the phenotypic variation of other proteins of interest and has revealed interesting sources of disease resistance in rice.
Introduction
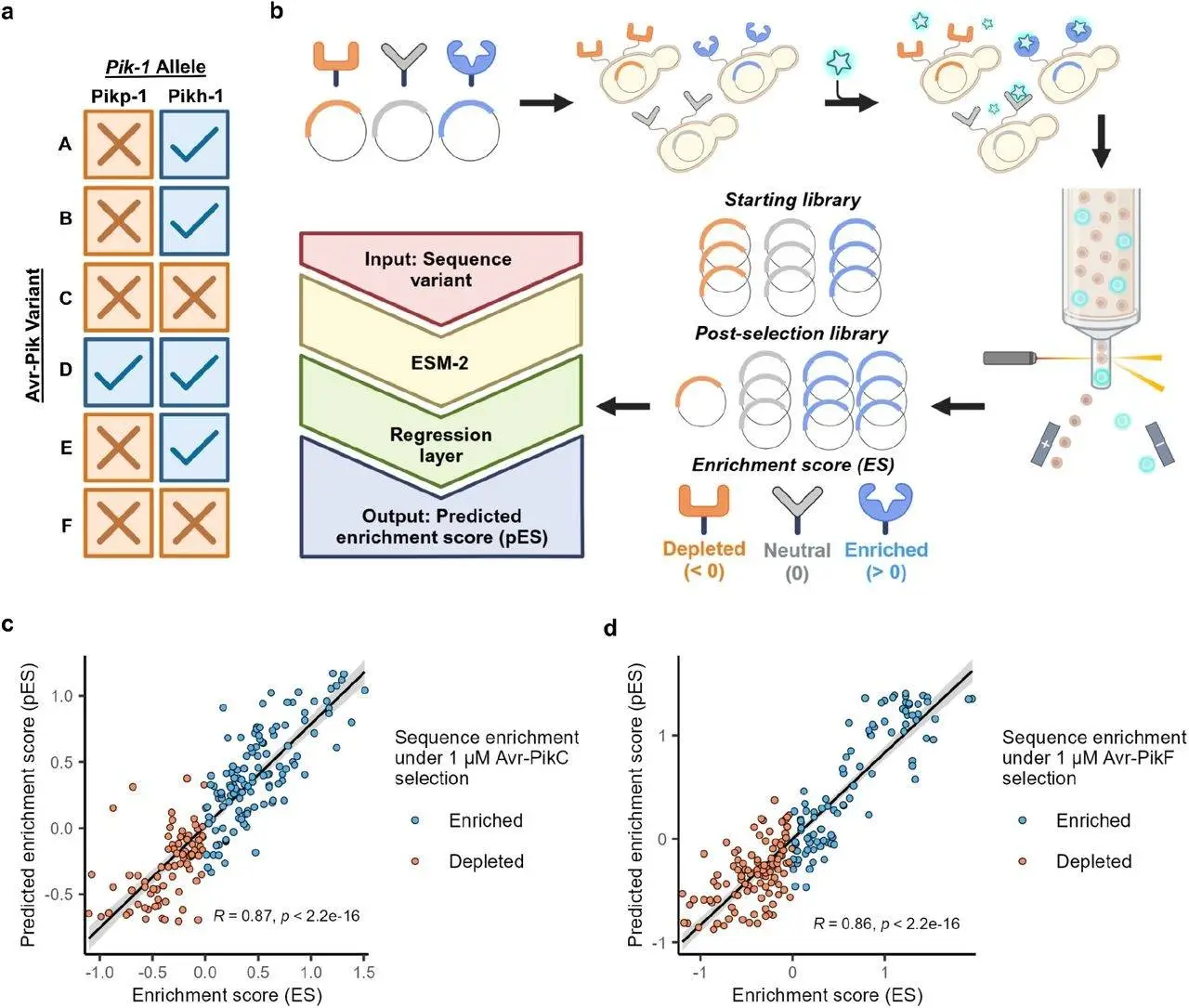
Neural networks trained on vast collections of evolutionarily developed proteins are known as protein language models (PLMs), and they are used to understand the sequence, structure, and functions of proteins. Input protein sequences can be reduced by these models into high-dimensional numerical representations known as embeddings, which have been applied to specific machine-learning applications. A more specific model that accurately forecasts the characteristics of an input protein sequence can be created by optimizing PLMs. Accurate predictions of how missense mutations may impact protein stability, enzyme function, and protein-protein interactions have been made using fine-tuned PLMs. Because of their accuracy and adaptability, fine-tuned PLMs are a helpful tool for forecasting how sequence variation may affect desired phenotypes. They are particularly beneficial when examining huge genomic datasets.
Exploring the Power of Machine Learning in Genomics
The study adjusted ESM-2 to predict Pik-1 HMA domain variant binding against Avr-PikC and Avr-PikF using directed evolution data. The relative change in sequence abundance between the initial and post-selection YSD libraries was quantified using the enrichment score (ES). The data was separated into training and validation sets to determine a projected enrichment score for input receptor variations. With a Spearman correlation coefficient R-value of more than 0.85, the final Avr-PikC and Avr-PikF fine-tuned models demonstrated a substantial link between sequence features and ligand binding alterations. Compared to other models trained on ESM-2 embeddings, this method fared better.
The Pikh-1 HMA domain variations in rice types were found using the 3,000 Rice Genomes Project (3k RGP). When the sequence was compared to a reference genome, 13 distinct HMA variations were found. Eleven of these have not been phenotypically described for their ability to bind to Avr-PikC or Avr-PikF. For Avr-PikC and Avr-PikF binding, the Pikh-1 and Pikp-1 alleles scored poorly; however, for Avr-PikC binding, ten obtained a positive pES value. The model consistently gave Pik-1 variants with an insertion in the center of the HMA domain a high score for Avr-PikC binding when it was trained on sequence data free of insertion or deletion alterations. Two variants were selected for downstream phenotyping because they had distinct insertions and significant pES values.
The ligand binding of the Pikh-1, VK, and SHZ-2 Pik-1 HMA domains against Avr-PikA and Avr-PikC at a concentration of 1 μM was examined. VK and SHZ-2 were next in line, with VK exhibiting better binding and SHZ-2 having the highest affinity, while Pikh-1 demonstrated the strongest binding.
To predict phenotypic effects, the human enzyme Nudix hydrolase 15 (NUDT15) underwent a mutagenesis scan. Each version was given a functionality score (FS) based on the stability and functionality assessments of the NUDT15 variant. By correctly associating differences in the NUDT15 sequence with the likelihood of cytotoxicity, the model enabled the development of optimized treatment strategies. The final refined model obtained an R-value of 0.76, indicating the efficacy of the methodology.
The thiopurine cytotoxicity-related NUDT15 variations were among the 14 that were scored using a refined model. All harmful missense substitutions and benign mutants were appropriately assessed by the model, with the exception of one cytotoxic variant (K33E). According to Suiter et al., three NUDT15 variants that had cytotoxic insertion/deletion mutations were not graded. Most in-frame deletion, duplication, and insertion mutants were rated as nonfunctional by the model, while substitution mutants were rated as functional.
According to the study, hitherto unobserved genotypic variations can be phenotyped using refined PLMs trained on directed evolution data. Only two alleles that have been found recently in wild rice varieties show substantial binding to the ligand, indicating that Pik-1 binding to Avr-PikC is uncommon in rice. The variety of Pik-1 HMA domain variations found in the 3k RGP dataset confirms earlier findings that M. oryzae-induced selection is promoting Pik-1 HMA domain diversification. This selection pressure is simulated by the study’s directed evolution methodology, which ESM-2 can utilize to precisely correlate naturally occurring sequence variation with alterations in ligand binding. The 3k RGP was used to identify two Pik-1 HMA domain variants, VK and SHZ-2, which showed improved binding to Avr-PikC in vitro compared to Pikh-1.
Conclusions
Transformer-based models have proven to be effective in precisely associating desired phenotypes with genetic variants. PLMs were utilized in this investigation to anticipate increased Avr-PikC binding to Pik-1 mutants with an insertion in the HMA domain. After training on a sequence dataset devoid of insertions or deletions, the model was still able to predict the detrimental effects of sequence insertions and deletions on the risk of thiopurine cytotoxicity. This precision implies that optimized PLMs can accurately assess how uncommon or undiscovered genotypic traits affect desired phenotypes. In addition to helping find immune receptor variations in rice with unique ligand recognition capabilities, the method may be used to detect other relevant traits, like patient drug sensitivity risk.
Article Source: Reference Paper
Disclaimer:
The research discussed in this article was conducted and published by the authors of the referenced paper. CBIRT has no involvement in the research itself. This article is intended solely to raise awareness about recent developments and does not claim authorship or endorsement of the research.
Important Note: bioRxiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Follow Us!
Learn More:




Deotima is a consulting scientific content writing intern at CBIRT. Currently she's pursuing Master's in Bioinformatics at Maulana Abul Kalam Azad University of Technology. As an emerging scientific writer, she is eager to apply her expertise in making intricate scientific concepts comprehensible to individuals from diverse backgrounds. Deotima harbors a particular passion for Structural Bioinformatics and Molecular Dynamics.











{kind=link}