
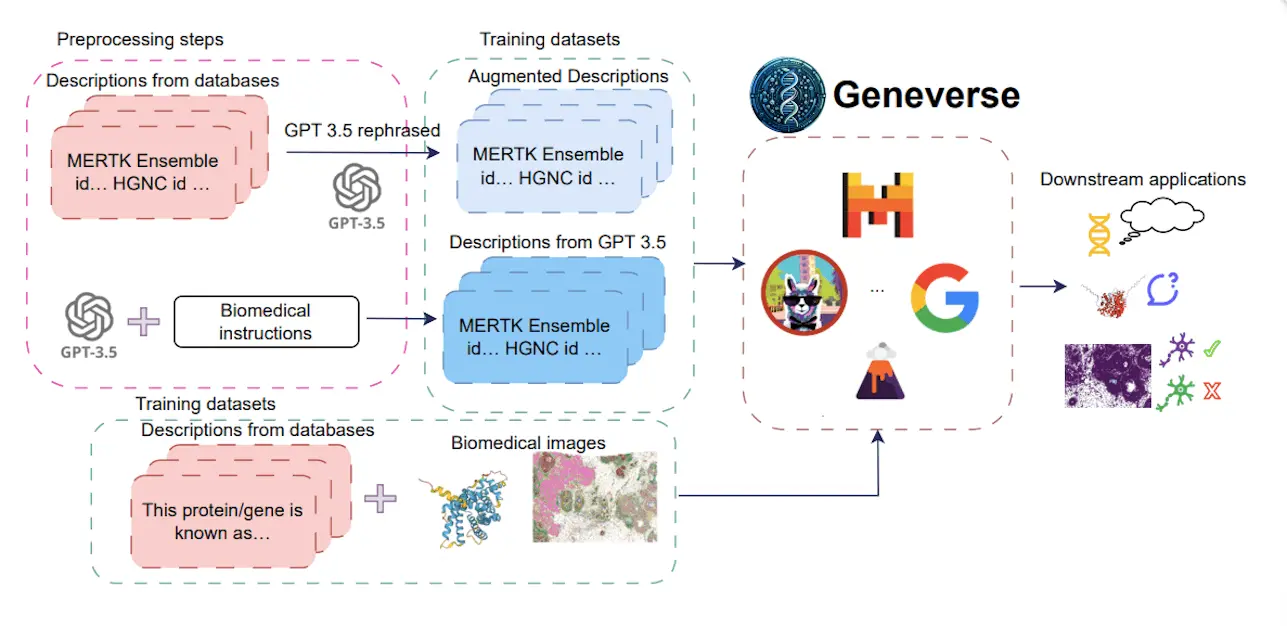
Scientists from Yale University, UCLA, and UT Health have introduced Geneverse, a collection of fine-tuned LLMs and multimodal LLMs (MLLMs) for genomic and proteomic research. These models address three novel tasks: generating gene function descriptions, inferring protein functions from structures, and selecting marker genes from spatial transcriptomic data.
Artificial intelligence (AI) is being adopted more frequently in genomics and proteomics science, revolutionizing the field. Large language models (LLMs) and powerful computer programs trained on huge amounts of text data have given encouraging results in several scientific domains. Although today’s LLMs can sum up information and generate text, they often fall short when applied to genomics and proteomics research with specific needs. Among these are that they must be accurate, compatible with established scientific knowledge, and understandable to various researchers.
Geneverse solves these problems through some key characteristics, such as fine-tuned LLMs based on existing open-source models; MLLMs include multimodal data and generally focus on the truthfulness and structure of outputs created by models. Geneverse could be useful for researchers in gene function prediction, protein function inference, or marker gene identification. Furthermore, it highlights various findings from the study, including the superior performance of Mistral-7B, the efficacy of using synthetic data augmentations, and the lackluster performance of RAG-based approaches.
Why Geneverse? Limitations of Existing LLMs
Although LLMs have the potential to summarize information and generate text, the current models often fail to deal with the specific needs of genomics and proteomics studies. These needs are:
Accuracy: Genomic and proteomic data require precise and verifiable knowledge. Mistakes in LLM outputs may lead to wrong final results.
Scientific Basis: The models should follow established scientific concepts for their predictions to be considered valid.
Accessibility: Many powerful LLMs are closed-source or require substantial computational resources, which narrows down their use by the wider scientific community.
Geneverse: Tackling the problems
The Geneverse approach towards these challenges is embodied in various ways:
Fine-tuned LLMs: This assemblage contains LLMs that are based on open-source models like LLaMA, Mistral, and Gemma. Moreover, such models go through fine-tuning with domain-specific datasets from databases like NCBI or synthetic data created with advanced language models such as GPT-3.5. The objective of the fine-tuning phase is to make these models meaningful and able to understand genomic and proteomic information.
Multimodal LMMS (MLMS): MLLMs are a step further in LLMs seen in Geneverse. This encompasses both textual data and visual information like protein structures and spatial transcriptomics.
Truthfulness vs Structure focus: Geneverse’s evaluation approach accords equal importance to truthfulness and structural correctness of model outputs, thereby ensuring that the explanations made and future predictions are not only scientifically correct but also logically organized for easy understanding by scientists.
Applications of Geneverse
Geneverse provides a toolkit for genomics and proteomics researchers who are handling different tasks.
These are a few examples:
Prediction of gene function: The descriptions generated on genes can be used by scientists to uncover their roles in biology.
Inference of protein function: MLLMs combine protein structure data with descriptions to achieve functional inference about a protein, which helps researchers understand the role of such a molecule within the cell.
Marker Gene Identification: MLLMs identify marker genes for particular cell types whose expression levels indicate this kind of cell. Cellular differentiation and development are important areas of study.
Results from Benchmarking and Main Findings
This study offers a comprehensive review that compares different LLM architectures and training strategies. Some significant outcomes include the following:
Mistral-7B is better than others: Of these models, Mistral-7B, an open-source LLM, performed best in terms of factuality, structurality, as well as conventional language model assessment metrics such as BLEU score or ROUGE score. It demonstrates how beneficial it is to fine-tune open-source LLMs for specific applications.
Data augmentation using synthetic data: Including GPT-3.5 generated synthetic data improved the generative outputs’ quality. This shows that Language Model LM is getting advanced enough to support augmentedness for better training outcomes.
RAG-based Methods Underperform: The use of Retrieval-Augmented Generation (RAG) was not ideal for these language models. They had no structure in their outputs, which sometimes contained irrelevant information.
Conclusion
Despite Geneverse’s achievements, the authors point out its shortcomings as well. A critical issue related to fine-tuning LLMs is the cost of computation involved, which necessitates the availability of powerful GPUs, but not all researchers will have access to them. This bottleneck in computing may limit how widely Geneverse can be adopted. Besides, a small number of tasks are considered as part of an evaluation at present. Further examination is required to encompass diverse applications pertinent to genomics and proteomics research so that one can fully comprehend all Geneverse abilities. At last, the second iteration of this tool is mainly meant for scientific study purposes only and might require future transformations for commercial or clinical purposes.
The researchers are currently addressing these limitations. They hope to optimize the training procedure to enhance model efficiency and minimize computational resources necessary for fine-tuning. Moreover, they intend to extend their evaluation into other common tasks faced in the genomics and proteomics field. Lastly, incorporating bigger and more diversified datasets might boost model performance and generalizability, hence making it useful across various research undertakings since it can be applied to wider spectrum efforts through gene verse generation itself.
Join the conversation
When it comes to the use of AI in genomic and proteomic research, Geneverse is doing an outstanding job. Geneverse is an open-source tool that emphasizes scientific correctness by special models. Moreover, this approach enables researchers to study biological data more efficiently to find new ideas.
Want to add something? What are your views concerning the influence of AI on genomics research?
Article Source: Reference Paper | The codes used in the study are available on GitHub.
Important Note: arXiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Follow Us!
Learn More:




Anchal is a consulting scientific writing intern at CBIRT with a passion for bioinformatics and its miracles. She is pursuing an MTech in Bioinformatics from Delhi Technological University, Delhi. Through engaging prose, she invites readers to explore the captivating world of bioinformatics, showcasing its groundbreaking contributions to understanding the mysteries of life. Besides science, she enjoys reading and painting.











 and multimodal LLMs for genomic and proteomic research.){kind=link}