

The DNA, RNA, and proteins that control an organism’s entire functioning are all fully encoded in a sequence called the genome. Large-scale genome databases and machine learning advances may make it possible to create a biological foundation model that speeds up the generative design and mechanistic analysis of intricate molecular interactions. Researchers from Arc Institute, TogtherAI, and collaborators present Evo, a genomic foundation model that allows for problems related to creation and prediction at both the molecular and genome scales. The deep signal processing architecture Evo has been scaled to 7 billion parameters at single-nucleotide byte resolution, with a context length of 131 kilobases (kb). Evo, having been trained on entire prokaryotic genomes, is capable of outperforming domain-specific language models in zero-shot function prediction. For the first time, it can create whole transposable systems and artificial CRISPR-Cas molecular complexes, demonstrating its proficiency in multielement production tasks. Evo has the potential to advance our knowledge and management of biology at many levels of complexity, as evidenced by its ability to predict gene essentiality at nucleotide precision and produce coding-rich sequences up to 650 kb in length.
Introduction
Rapid advances in DNA sequencing technologies have made it possible to map evolutionary variation systematically at the whole-genome scale, exposing the basic biological information that drives evolution’s generational transmission. This diversity is a result of selection and adaptation for phenotypic biological function. A broad biological foundation model that learns the underlying logic of entire genomes—thereby permitting a thorough comprehension of intricate biological processes and disease mechanisms—may be made possible by the combination of machine learning techniques with enormous genomic sequence databases.
Molecular biology is being modeled using machine learning, with an emphasis on proteins, regulatory DNA, and RNA. On the other hand, a DNA model that integrates information across molecular systems and genome sizes is necessary for complicated biological processes, including genetic transposition, CRISPR immunity, and gene control. Large genomic regions might be learned from this model, which could also capture connections between systems and facilitate the construction of more complex biological activities. This model incorporates the evolutionary implications of sequence variation, such as specific single-nucleotide mutations that change organism function by functioning at single-nucleotide resolution.
Inspired by recent successes, transformers have been utilized in language models to capture biological sequences and system-wide interactions. Nevertheless, the dense Transformer architecture, which is computationally expensive and works poorly at the single-nucleotide or byte-level resolution, poses a barrier to current efforts to describe DNA as a language. Transformer-based DNA models are limited to small context lengths and use tokens to aggregate nucleotides, a restriction also present in recent developments in attention-based models.
Introducing Evo
A genomic foundation model with 7 billion parameters, Evo is taught to produce DNA sequences at the whole-genome level. The StripedHyena architecture is utilized to process and recall patterns in lengthy sequences effectively by combining data-controlled convolutional operators and attention. Evo employs a byte-level, single-nucleotide tokenizer and is trained on a 300 billion nucleotide bacterial whole-genome dataset. Evo predicts combinations of prokaryotic promoter-ribosome binding site pairs that result in active gene expression from regulatory sequence alone, outperforms specialized RNA language models in predicting the fitness effects of mutations on non-coding RNAs, and is competitive with state-of-the-art protein language models in zero-shot evaluations. Evo can produce sequences longer than 650 kilobases (kb) with a believable genomic coding architecture and autonomously predict key genes in bacteria or bacteriophages. Evo’s further growth will speed up our ability to build life and allow a deeper mechanistic understanding of biology.
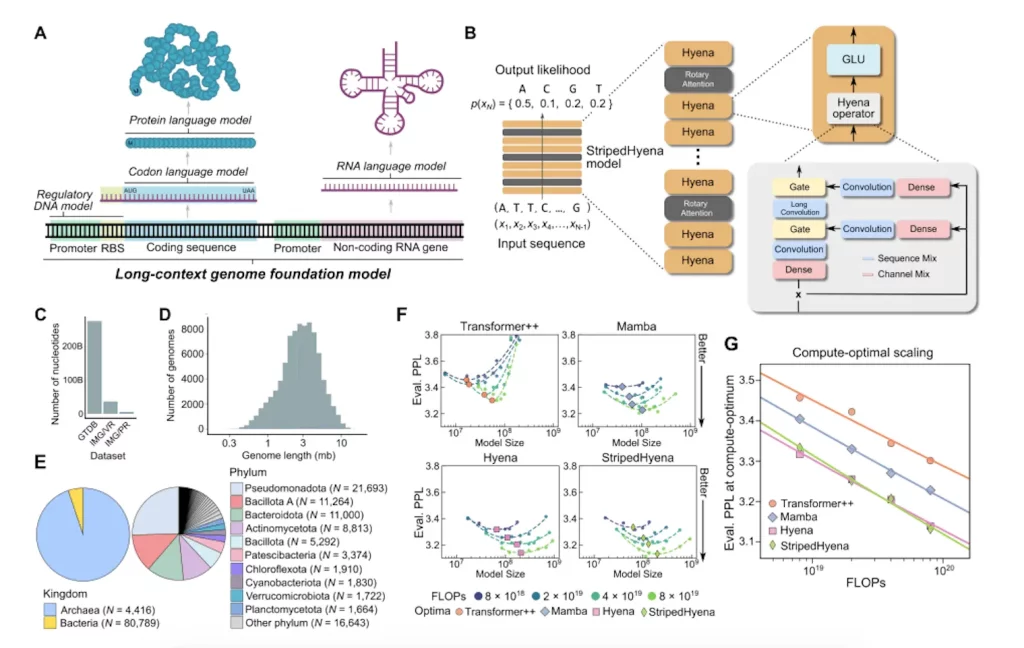
Image Source: https://doi.org/10.1101/2024.02.27.582234
Applications of Evo
- Estimating the impact of mutations on protein function – Without task-specific supervision or fine-tuning, Evo, a protein language model trained on lengthy genomic sequences containing protein-coding regions, has shown an excellent capacity to anticipate the effects of mutations on protein function. Deep mutational scanning (DMS) investigations were used to assess Evo’s zero-shot performance on downstream tasks that are biologically relevant. Top protein-specific language models were surpassed by Evo, which also beat all other examined nucleotide models, including GenSLM. However, Evo has not been trained on human protein DMS datasets, most likely due to the pretraining dataset including only bacterial sequences. Further optimization or pretraining on mammalian coding sequences should enhance Evo’s performance beyond bacterial proteins, as there is a strong correlation between language-model perplexity on the wildtype sequence and fitness prediction performance.
- Estimating the impact of mutations on ncRNA function – To discover useful knowledge about non-coding RNAs (ncRNA), including tRNAs, rRNAs, and ribozymes, Evo was put to the test. Mutations to the ncRNA sequence were present in the ncRNA DMS datasets used to train the model. Based on experimental ncRNA DMS results, Evo’s capacity for zero-shot ncRNA fitness prediction was assessed. As demonstrated by the results, Evo performed better than every other nucleotide language model that was examined, including RNA-FM, an RNA language model that was trained using non-coding RNA sequences. In research examining how mutations in the 5S ribosomal RNA affect the growth rate of Escherichia coli, Evo showed excellent predicting ability. It appears from these findings that Evo can anticipate functional features across several molecular modalities by using training data from its prokaryotic genome.
- Regulatory DNA-based gene expression prediction – It has been discovered that the prokaryotic model Evo, which was trained on regulatory DNA sequences, can predict gene expression. The likelihood ratings of the model predict binarized protein expression and have a significant correlation with mRNA expression. Since none of the nucleotide language models have been trained on datasets containing regulatory components, Evo has a stronger prediction performance than the others. Based on natural variation in protein sequences, the model probably predicts functional changes using its pretraining knowledge of natural regulatory sequences. Evo shows a grasp of constitutive protein-coding sequences, ncRNA sequences, and regulatory regions even though it was trained on lengthy genomic crops without precise sequence annotations.
Source: https://arcinstitute.org/news/blog/evo
Conclusion
Evo is a genomic foundation model that can predict and generate DNA sequences at the scale of individual molecules, chemical complexes, biological systems, and entire genomes. It was trained using hundreds of billions of DNA tokens from prokaryotic life. Evo, which is based on StripedHyena, allows for language modeling at a context length of 131k with single-nucleotide resolution. Evo matched or outperformed specialized models in accurately completing zero-shot prediction across a variety of fitness or expression prediction tasks on proteins, ncRNAs, or regulatory DNA. Still, in order to progress with medicinal discoveries, sustainability and a basic understanding of biology, biosafety, and ethical issues need to be taken into account. The features of systems biology and the prediction of molecular structures may both benefit from this paradigm. Additionally, Evo might serve as the foundation for a next-generation sequence search method that would allow for semantic or relational metagenomic mining. However, eukaryotic genome incorporation necessitates a significant investment in computing, safety-related model alignment, and engineering.
Article source: Reference Paper | Reference Article | Code and models related to this study are publicly available on GitHub
Important Note: arXiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Follow Us!
Learn More:




Deotima is a consulting scientific content writing intern at CBIRT. Currently she's pursuing Master's in Bioinformatics at Maulana Abul Kalam Azad University of Technology. As an emerging scientific writer, she is eager to apply her expertise in making intricate scientific concepts comprehensible to individuals from diverse backgrounds. Deotima harbors a particular passion for Structural Bioinformatics and Molecular Dynamics.











{kind=link}