
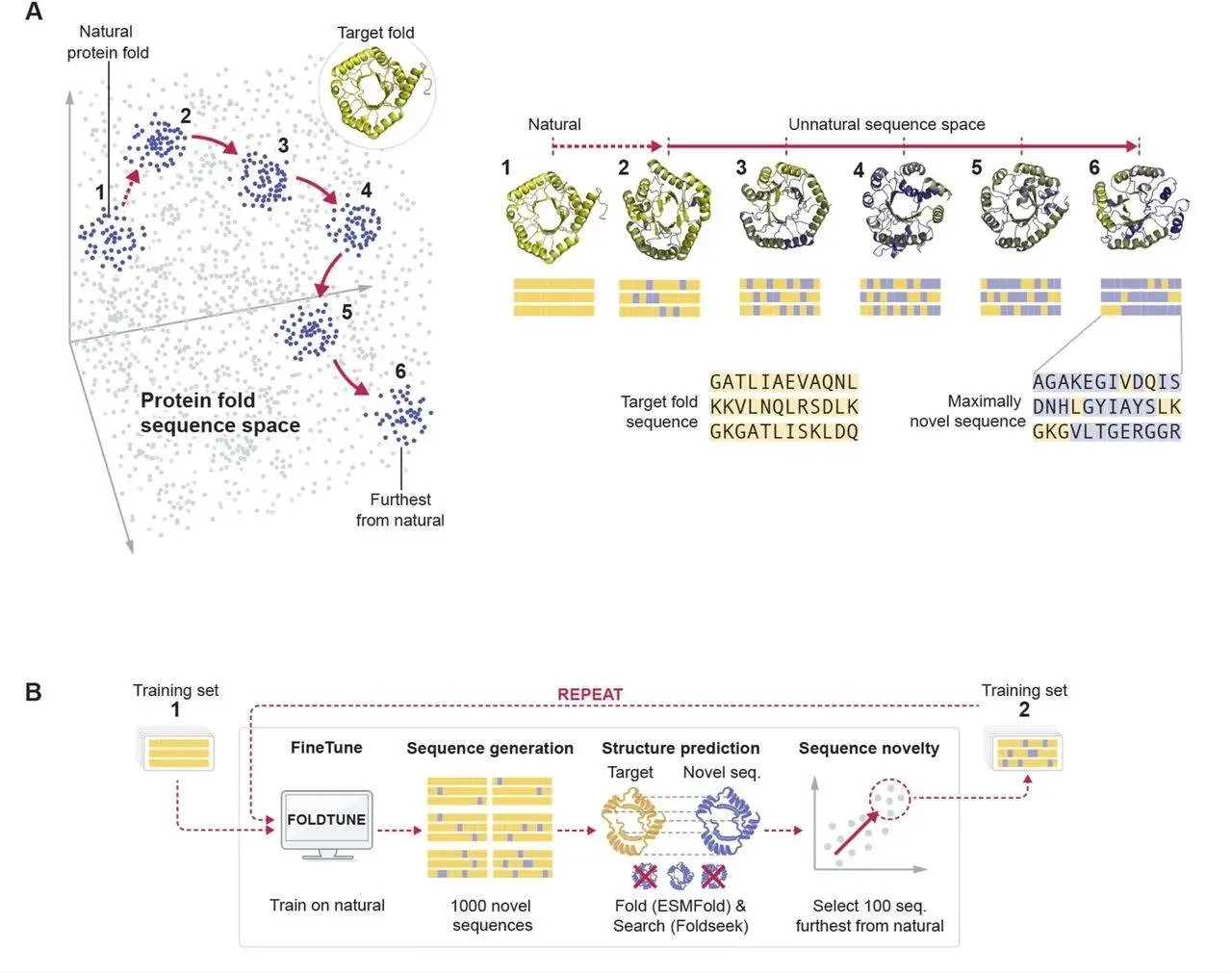
The study of the entire protein sequence-structure map has historically been hampered by the combinatorial scale of amino acid sequence space. The wide unexplored landscape of far-from natural sequences that appear to have escaped evolution but are consistent with the principles of biophysics is not yet known. Experimental methods such as directed evolution are not able to reach these spaces due to the scale of sequence perturbations needed. By using generation and self-feedback, researchers overcome this restriction, which is driven by the inherent ability of protein language models (pLMs) to investigate sequences that are not included in their natural training set. In a novel method to sequence design that researchers from the California Institute of Technology called “foldtuning,” they recast pLMs as probes that explore areas of protein “deep space” that have little to no apparent homology to natural instances while enforcing key structural constraints. Researchers create a library of foldtuned pLMs for over 700 natural folds in the SCOP database, encompassing many high-priority targets for synthetic biologies, such as small GTPases and GPCRs, DNA-binding and composable cell-surface receptors, and tiny signaling/regulatory domains.
Introduction
Nature probably only sampled a small portion of the protein shapes and sequences permitted by the principles of biophysics. One hundred nine distinct protein sequences are spread throughout the Tree of Life, ensuring a combinatorially large sequence space thanks to the 20 proteinogenic amino acids. One hundred nine distinct protein sequences are found in high-quality sequence databases; these sequences were probably chosen based on criteria including advantageous folding kinetics, cofactor utilization, and binding/catalytic activities. Nevertheless, hydrophobic/polar patterning approaches produce stable U-helical bundle proteins encoded by new sequences and differentiate energetically favorable three-dimensional structures.
Deep multiple sequence alignments can capture enough sparse co-evolutionary signals to produce synthetic proteins that are as stable as those found in nature. Random sequence library measurements indicate that up to 1 in 1011 amino acid sequences might code for functional proteins, offering plenty of “sparks” for protein populations outside of nature. The systematic identification of stable, functional proteins that reassemble known structural motifs but are located in sequence-space regions that lack significant natural resemblance holds promise for expanding synthetic biology’s repertoire of binding partners, signaling interactions, and substrate scopes while exposing important amino acid sequence constraints and rules that underlie the basic biophysics of molecular machines.
Foldtuning of Language Models for Protein Mapping and Many More
The difficulty of mining “doppelganger” proteins can be mitigated by protein language models (pLMs), which strike a balance between minor backbone structure alterations and significant sequence disruptions. Whereas inverse-folding structure-to-sequence design techniques can diversify sequences while enforcing stringent backbone restrictions, directed evolution and machine learning models are not appropriate for managing global sequence disturbances. Protein language models automatically absorb the information flow from sequence to structure by explicitly learning amino acid relationships at the sequence level. As protein producers, pLMs are capable of going beyond the sequences and structures found in nature.
The method introduced in this study, “foldtuning,” turns pLMs into probes that follow structure-preserving routes through distant areas of protein sequence space since pLMs comprehend the fundamental factors of sequence-to-structure mapping while maintaining an intrinsic exploratory ability.
Foldtuning searches protein space for examples that respect the sequence “grammar” of a target backbone piece while displaying unique semantics by utilizing the complimentary properties of pLMs, structure prediction models, and ultrafast structure-based search techniques.
The SCOP and InterPro databases contain over 700 structural motifs of interest for which researchers have successfully applied foldtuning. These motifs span all four major tertiary topology classes and a wide range of functional families, such as transcription factors, GPCRs, cell-to-cell signaling domains, and different cytokines. The findings demonstrate that, in general, foldtuning cycles gradually decrease the similarity between pLM-generated and wild-type sequences, leading to the discovery of novel “rules of language” for protein construction while concurrently implementing small structural modifications like loop expansion/minimization and symmetry adaptation.
Foldtuned sequence libraries for two target folds—the SH3 domain, which regulates tyrosine kinase and small GTPase, and the barstar ribonuclease inhibitor—are screened in high-throughput using mass spectrometry-based proteomics and survival assays, respectively. This finds stable, functional candidate variants with 0–40% sequence identity to their closest neighbors in the known protein universe. At the end of the day, foldtuning, or sequence remodeling, pushes the boundaries of how much structure and function can be preserved while diversifying typical protein folds at the sequence level.
Conclusion
The goal of the foldtuning technique is to locate and track putative protein signals in amino acid sequence space. Foldtuned models suggest significant changes to the protein sequence while preserving minor changes to the target backbone structure for several hundred target folds by building a library of ProtGPT2 variants. In local and global sequence settings, these sequences display patterns of usage that are strange and far from normal. Tested foldtuned protein variations exhibit properties of stable, well-folded, and useful synthetic proteins, pointing to novel binding and recognition mechanisms and directing future advancements in generating capacity. Since the foldtuning procedure is modular, it is possible to make changes like combinatorial diversification of protein domains and subdomains, end-to-end model updates, and substituting scoring techniques specific to the engineering challenge for the computationally demanding structural validation stage.
Article Source: Reference Paper
Disclaimer:
The research discussed in this article was conducted and published by the authors of the referenced paper. CBIRT has no involvement in the research itself. This article is intended solely to raise awareness about recent developments and does not claim authorship or endorsement of the research.
Important Note: bioRxiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Follow Us!
Learn More:




Deotima is a consulting scientific content writing intern at CBIRT. Currently she's pursuing Master's in Bioinformatics at Maulana Abul Kalam Azad University of Technology. As an emerging scientific writer, she is eager to apply her expertise in making intricate scientific concepts comprehensible to individuals from diverse backgrounds. Deotima harbors a particular passion for Structural Bioinformatics and Molecular Dynamics.











{kind=link}