
Shotgun metagenomics has revolutionized microbiome research, allowing scientists to study microbial communities at an unprecedented depth. However, analyzing the massive amounts of sequencing data generated by this technique is no easy task. To address this challenge, researchers from multiple institutions across China specializing in microbiome research, bioinformatics, and genomics have developed EasyMetagenome, a user-friendly and flexible pipeline designed to streamline metagenomic analysis.
What is EasyMetagenome?
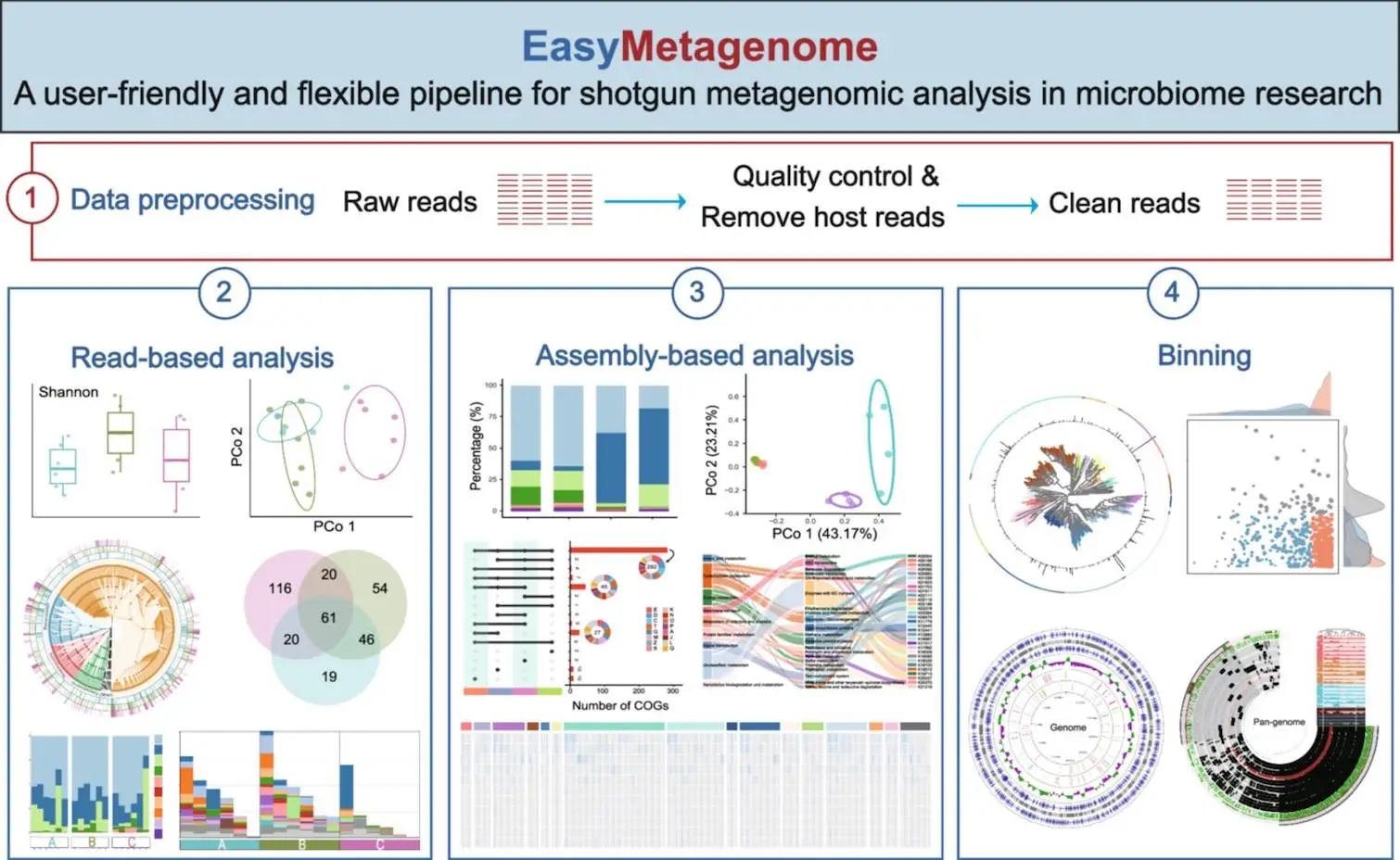
EasyMetagenome is a free-to-use platform that facilitates the analysis of the sequence data obtained from a metagenomic sample. It encompasses quality filtering, function and taxonomic profiling, genome assembly, metagenome binning, and microbiome integration in a comprehensive framework.
This versatility is one of the main strengths of EasyMetagenome. Users can adjust and even remove nearly all steps in the workflow depending on their data and the scope of research. Moreover, the analysis can be performed with just the default values for the parameters, which will maximize the ease of use and speed of implementation. Furthermore, intelligent visualizations are produced and make publication without additional steps simple.
Key Features of EasyMetagenome
The EasyMetagenome package has four major components:
- Software and Database Installation – The pipeline is equipped with 150+ bioinformatics tools and libraries corresponding to different steps of metagenomic analysis. It also has several installation options, which makes it adaptable for users with different computer systems.
- Data Analysis Pipeline – The most important part of EasyMetagenome is the read assembly and binning analyses, which allow users to investigate the microbial communities in all aspects.
- Statistics and Visualization – The provided R scripts help users draw different and more complex statistics and figures for easier interpretation of the provided data.
- Dedicated Q&A Section – This section helps the users by providing tips for parameter settings and troubleshooting, which enhances the user’s ability to control their analyses.
How Does EasyMetagenome Work?
- Data Preprocessing: The first step is performing quality control and host genome removal. For this, fastp and KneadData are employed to perform read quality filtering and non-microbial sequence degradation to ensure clean input data.
- Read-Based Analysis: Preprocessed data can now be analyzed for taxonomic composition and functional annotation with Kraken2 and HUMAnN3, respectively. The former is used for taxonomic classification, and the latter maps reads to known functional pathways. This method offers wide coverage of the microbial community but is likely to miss novel genes that are absent in the current reference databases.
- Assembly-Based Analysis: To dig deeper, EasyMetagenome allows users to assemble reads into longer sequences (contigs) using MEGAHIT or metaSPAdes. The assembled contigs are subsequently predicted in terms of genes and have their functions annotated to correlate microbial species with associated metabolic functions.
- Metagenome Binning: In this step, assembled contigs are clustered into Metagenome Assembled Genomes (MAGs) with the help of MetaWRAP. This approach enables the genome reconstruction of single microbes to understand their evolutionary relationships and functional potential.
- Data Visualization: EasyMetagenome includes built-in R scripts for statistical analysis and visualization. Users can generate taxonomic bar plots, functional heatmaps, and phylogenetic trees, making it easier to interpret complex microbiome datasets.
Why Choose EasyMetagenome?
EasyMetagenome serves as an intermediatory pipeline for metagenomic research inclined towards flexibility and user-friendliness. They make installation and usage smooth, which helps researchers with little to no experience in bioinformatics. It also supports both short-read and long-read sequencing data, which makes it useful in a multitude of studies. It is community-driven and open-source, which helps in fostering collaboration and improvement.
The range of applications makes it a great tool for research in identifying microbial signatures related to health and disease in the human gut microbiome, environmental microbiology to study microbial communities in different ecosystems, detection of resistance genes in pyogenic microorganisms for antibiotic resistance research, and soil, plant and fermented food microbiome in food and agriculture.
The Future of EasyMetagenome
The developers of EasyMetagenome seek to provide more functionality by embedding AI for microbiome analysis, improving the compatibility with 3rd generation sequencing technologies, and solving host contamination complicacies in microbiome data.
As metagenomic research continues to grow, tools like EasyMetagenome will be essential for unlocking the full potential of microbiome data and driving new discoveries in microbiology, health, and environmental sciences.
Article Source: Reference Paper | EasyMetagenome is freely available on GitHub.
Disclaimer:
The research discussed in this article was conducted and published by the authors of the referenced paper. CBIRT has no involvement in the research itself. This article is intended solely to raise awareness about recent developments and does not claim authorship or endorsement of the research.
Follow Us!
Learn More:




Anchal is a consulting scientific writing intern at CBIRT with a passion for bioinformatics and its miracles. She is pursuing an MTech in Bioinformatics from Delhi Technological University, Delhi. Through engaging prose, she invites readers to explore the captivating world of bioinformatics, showcasing its groundbreaking contributions to understanding the mysteries of life. Besides science, she enjoys reading and painting.











{kind=link}