
The inability to logically design new antibodies that bind a particular epitope on a target persists despite the crucial role that antibodies play in contemporary medicine. Instead, time-consuming animal immunization procedures or library screening techniques are currently used in antibody development. Here, scientists from the University of Washington show that user-specified epitopes can be bound by de novo antibody variable heavy chains (VHHs) created by a refined RFdiffusion network. Researchers have confirmed binders to four disease-relevant epitopes through experiments, and the overall binding pose and CDR loop configuration of a proposed VHH bound to influenza hemagglutinin in the cryo-EM structure are almost the same as in the design model.
Introduction
Protein therapies, of which antibodies are the predominant class, now have over 160 licenses worldwide; in the next five years, the industry is projected to reach $445 billion. Therapeutic antibody development frequently depends on animal immunization or antibody library screening despite the great interest in these antibodies from the pharmaceutical industry. In addition to being time-consuming and difficult, these techniques may not provide antibodies that bind with the therapeutically important epitope. Several techniques have been used in the computational design of antibodies, such as utilizing the Rosetta sequence design, sampling different native CDR loops, and grafting residues onto pre-existing structures. De novo design of structurally correct antibodies has been difficult to achieve, though. Designing binding proteins with RFdiffusion has made it possible to create a wide variety of binders that are naturally shaped to complement the user-specified epitope. However, RFdiffusion is unable to design antibodies de novo.
RFdiffusion and RoseTTAFold2, two methods, are being developed to design de novo antibodies. The approach involves:
- Targeting any epitope on any target.
- Focusing on CDR loops.
- Sampling alternative rigid-body placements.
The underlying thermodynamics of interface formation remain the same, allowing for the development of specialized versions capable of designing de novo antibodies. RoseTTAFold2 and RFdiffusion are trained on the entire Protein Data Bank (PDB), which helps overcome the problem of the PDB having relatively few antibody structures. The aim is to develop versions of RFdiffusion and RoseTTAFold2 specialized for antibody structure design and structure prediction by fine-tuning native antibody structures. The original RFdiffusion network is referred to as “vanilla RFdiffusion,” while the antibody-specific variant is simply “RFdiffusion.”
Looking Into RFdiffusion
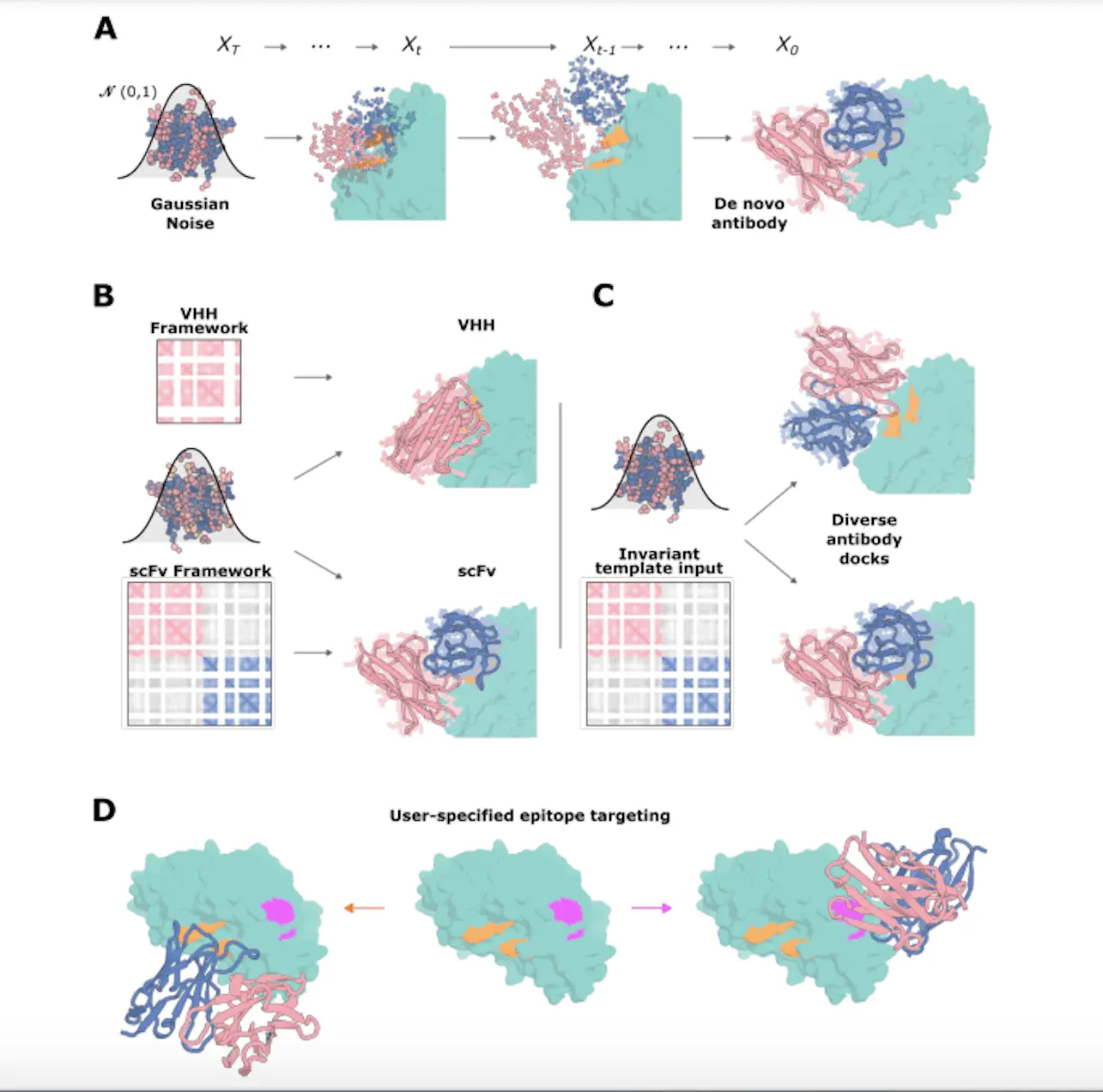
RFdiffusion is a training method that uses the AlphaFold2/RF2 frame representation of protein backbones, including residue orientations. Protein frames are distorted over timesteps by means of a noising schedule, which leaves distributions that are identical to random distributions. Training involves sampling a PDB structure, applying t-noising steps, and selecting a random timestep (t). Predicting the de-noised structure at every timestep, RFdiffusion minimizes the mean squared error loss between the predicted and the genuine structure. In order to produce a novel protein structure, translations are denoised at inference time by sampling from 3D Gaussian and uniform rotational distributions.
Enhancing RFdiffusion
Antibody complex structure tuning is the main goal of the training technique known as RFdiffusion. When training, a random timestep (t) is added to an antibody complex structure sample in order to corrupt the antibody structure but not the target structure. To facilitate the specification of the framework structure and sequence at the time of inference, RFdiffusion is trained with the framework sequence. A global-frame-invariant framework structure is supplied, and the internal structure of every protein chain is specified by the target template and the framework. Targeting the designated epitope with unique CDR loops, the training regime enables RFdiffusion to create antibody structures that nearly resemble the input framework structure. ProteinMPNN is used to design the CDR loop sequences, and the designed antibodies make diverse interactions with the target epitope, differing significantly from the training dataset.
RoseTTAFold2’s fine-tuning for antibody design validation
An improved filter has been developed for structure prediction of antibody structures by fine-tuning the RoseTTAFold2 network. While precisely modeling the CDRs and calculating the direction of the antibody against the targeted region, this network also offers information about the target structure and target epitope. Using this method, RF2 is able to recognize genuine antibody-antigen combinations from bogus pairs and make precise predictions about complicated structures. A useful tool for predicting antibody-antigen complex structures, the fine-tuned RF2 surpasses the previously published IgFold network in monomer prediction, especially in CDR H3 structure prediction.
According to Rosetta ddG measurements, RFdiffusion-designed VHHs are expected to bind nearly exactly like the intended structure, with a sizable portion of them creating high-quality interfaces. It is seldom predicted that these VHHs will bind to unrelated proteins, according to in silico cross-reactivity analyses. The observation that RF2 predicts a large number of designed sequences to adopt the designed structures and binding modes raises the possibility that RF2 filtering could enhance experimentally successful binders.
Design of VHHs
The goal of the research is to develop single-domain antibodies (VHHs) by using the variable domain of heavy-chain antibodies made by sharks and camelids. Comparing VHHs to fragment antigen-binding regions (Fab) or single chain variable fragments (scFv), the former are more affordable and simpler to build. The average interaction surface area of a VHH is comparable to that of an Fv, even though it has fewer CDR loops, indicating that a technique that works well for designing VHHs should work well for designing Fvs as well. Rosetta and optimized RF2 were employed to evaluate the interface characteristics.
The work shows that VHHs that specifically bind with the target epitope, like influenza HA, along the HA-stem epitope may be designed using RFdiffusion. The shortened paucimannose glycan shield expressed by insect cells is similar to the fully deglycosylated HA monomeric PDB model that was utilized in the creation of VHH. The most highly affinitized binders to TcdB, Influenza HA, Covid RBD, and RSV site III were discovered. Additionally, the statistics demonstrate that using the RF2 settings, the design success rates for filtered designs compared to unfiltered designs were not substantially higher. Larger datasets are required to determine how effectively to use and fine-tune RF2 for design filtering.
Conclusion
The extremely variable H3 loop known as the VHH complex was created through the use of RFdiffusion, a computational de novo design technique. Compared to immunizing animals or screening random libraries, this method is quicker and less expensive because it enables the selective targeting of particular epitopes of interest on the target antigen. The structure-based approach to antibody design prevents mutations that could destabilize the antibody or disturb the antibody-target interface while optimizing important medicinal qualities, including aggregation, solubility, and expression level. The optimization of developability features and targeting non-immunodominant epitopes is made easier by exploring the entire space of CDR loop sequences and structures, especially for CDR1 and CDR2. Additionally, every antibody designed by RFdiffusion has a strong structural hypothesis, enabling the rational design of antibody function by targeting specific target conformational states.
Despite the effectiveness of VHH design, low success rates and modest binding affinities provide space for development. Higher designability and diversity design models may result from recent architectural advancements or from more recent generative frameworks like flow matching. By modeling all biomolecules, including glycans, epitopes comprising non-protein atoms, roseTTAFold2, and vanilla RFdiffusion may now be used to build antibodies against them. While ProteinMPNN was left unaltered in this work, the potential immunogenicity of developed antibodies could be mitigated by constructing sequences that closely match human CDR sequences. Future work may also focus on directly improving ProteinMPNN’s developability attributes. Better in-silico benchmarking of upstream design methods and higher experimental success rates could be possible with further advancements in RoseTTAFold2 antibody prediction techniques.
Article source: Reference Paper | Reference Article
Important Note: bioRxiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Follow Us!
Learn More:




Deotima is a consulting scientific content writing intern at CBIRT. Currently she's pursuing Master's in Bioinformatics at Maulana Abul Kalam Azad University of Technology. As an emerging scientific writer, she is eager to apply her expertise in making intricate scientific concepts comprehensible to individuals from diverse backgrounds. Deotima harbors a particular passion for Structural Bioinformatics and Molecular Dynamics.











 to design single-domain antibodies from scratch with atomic precision.){kind=link}