
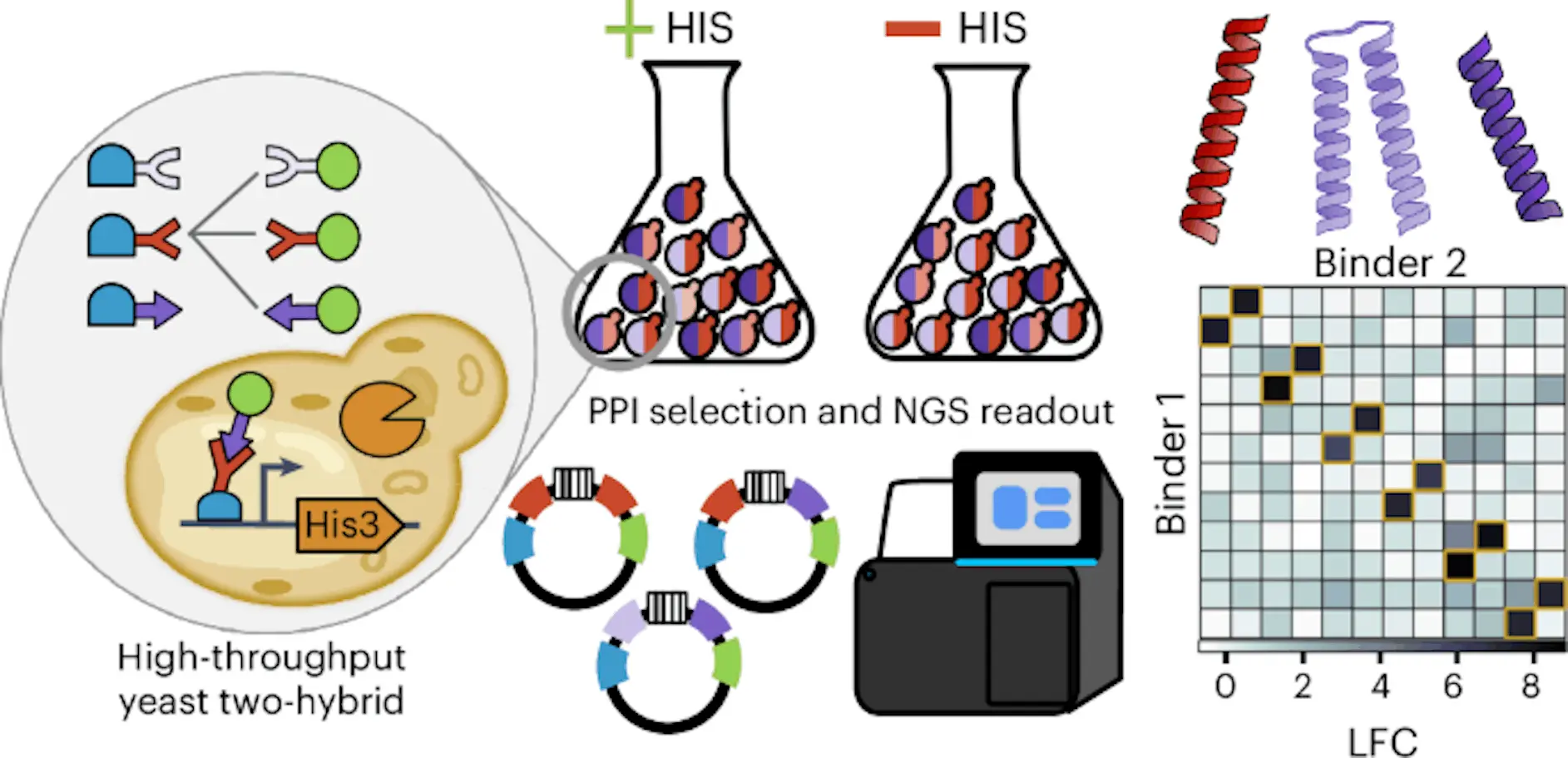
Numerous biological processes are regulated by protein-protein interactions (PPIs), and modified PPIs have uses in gene and cell therapy. Here, researchers from the University of Washington present a simple, highly scalable yeast two-hybrid method for assessing PPIs called massively parallel PPI measurement by sequencing (MP3-seq). DNA barcodes in MP3-seq are linked to particular protein pairs, and barcode enrichment can be interpreted by sequencing to yield an accurate assessment of the strength of the connection. Researchers demonstrated that MP3-seq scales to over 100,000 interactions and is quite quantitative. Researchers use MP3-seq to study factors granting specificity to coiled-coil interactions and to characterize interactions between families of logically formed heterodimers. Finally, researchers use AlphaFold-Multimer (AF-M) to predict coiled heterodimer structures and train linear models on physics-based energy terms to predict MP3-seq results. Researchers conclude that while AF-M-based models may be useful for prescreening interactions, quantitative ranking of interaction strengths will still require experimental measurement of interactions.
Introduction
Potential revolutions in domains such as cell treatment, synthetic biology, and material science could be brought about by artificial protein binders that mediate interactions with other proteins or cells. Natural interaction domains like SH3 and PDZ were frequently used in earlier work. Unfortunately, due to crosstalk, such components offer a subpar beginning point for logically developing large-scale assemblies. As such, artificial protein circuits continue to be far smaller and less complex than biological protein-protein interaction (PPI) networks. Scientists require large-scale libraries of modular interaction domains in order to build up synthetic protein-based circuits. These interaction domains should ideally be orthogonal, meaning that each domain exclusively communicates with its assigned binding partner.
Large, fully orthogonal sets are still difficult to create, despite the encouraging achievements of rational heterodimer design, which has mostly focused on coiled-coil dimers (1×1s). Coil-coil dimers were the subject of early rational design studies. In these dimers, complementary hydrophobic and electrostatic interactions at particular heptad locations determine 1×1 coil binding. It has been feasible to produce orthogonal sets of up to six 1×1 heterodimers; however, the number of viable orthogonal contacts is limited by the restrictive geometry needed for 1×1 coil interactions. Higher-order orthogonal sets may be produced by using helical bundle heterodimers, as demonstrated by the Baker lab. As these are not included in the biophysical design goal to generate 2×2s, minimizing off-target interactions is still difficult. A useful substitute would be to create several de novo 2×2 protomer libraries and quantify an all-by-all interaction matrix that would enable the orthogonal set to be extracted.
PPI Measuring Methods
Different throughput levels can be achieved by running all-by-all PPI displays using the numerous PPI measuring techniques that have been developed. While protein purification operations are time-consuming, mass spectrometry- and protein array-based techniques are capable of measuring PPIs. The “several-versus-many” screening that is possible with yeast-display or phage-display approaches is restricted to using next-generation sequencing technologies to boost throughput. Yeast mating pathway-based high-throughput screening technique called alpha-seq circumvents this restriction and enables library-on-library screening. All proteins do not, however, fold appropriately when exhibited on the yeast surface, thus throughput is still restricted.
What is Yeast Two-Hybrid (Y2H)?
Yeast two-hybrid (Y2H) methods are a powerful substitute for surface display for characterizing protein-protein interactions (PPIs). Using these techniques, two proteins are fused together: one to a transcriptional activation domain (AD), which is reconstituted when proteins interact, and the other to a DNA-binding domain (DBD). To overcome scaling problems and read out interaction strength, high-throughput Y2H (HT-Y2H) and enzyme complementation techniques have been developed. Additionally, specialized procedures for HT-Y2H data analysis have been created. The majority of experimental techniques necessitate the production of protein libraries in Escherichia coli or rely on yeast mating, which calls for distinct protein libraries. An alternative noneukaryotic method for screening PPIs without yeast mating and library transfer is the development of high-throughput bacterial two-hybrid tests. In comparison to bacterial expression, Y2H-based assays allow for more accurate folding, post-translational modifications, and higher solubility, making them useful for investigating interactions of naturally occurring intracellular proteins with eukaryotic origins.
Understanding MP3-seq
In this article, researchers present MP3-seq, a massively parallel Y2H pipeline for sequencing-based PPI measurement. Each protein in MP3-seq has its identity stored in a DNA barcode, and the relative abundance of barcode pairs before and after a selection experiment acts as a surrogate for interaction intensity. Yeast homologous recombination is used to build plasmids that encode the desired protein pairs, their corresponding barcodes, and all other components needed for Y2H research. The process folds and interacts with proteins inside the cell rather than on its surface, and it does so without requiring plasmid cloning in E. Coli or yeast mating. Unlike tests using microorganisms, Glycosylated proteins are likewise compatible with MP3-seq.
Using synthetic binders for several members of the human B cell lymphoma 2 (Bcl-2) protein family and well-characterized coiled-coil heterodimer interactions, scientists validate MP3-seq. Next, MP3-seq is employed to analyze interactions between logically constructed 2×2 and 1×2 heterodimers, proving that MP3-seq is capable of measuring more than 100,000 PPIs in a single experiment. Scientists found designs that worked and then used a greedy approach to locate subsets that might be orthogonal. By screening variants of a successful 2×2 pair, scientists investigated the components anticipated to impart specificity for 2×2 interactions. In conclusion, researchers forecast complexes for coiled-coil dimers including AF2 and AF-M, and evaluate the predictive capacity of these models for orthogonality and interactions. Researchers next train simple models to predict MP3-seq results using physics-based structural energy components from Rosetta and AF-M error values. This allows them to explore how complicated predictors could complement high-throughput data.
Conclusion
An easy-to-use technique for quantifying paired PPIs in a single yeast strain without surface display is the MP3-seq process. To combine replicates, exclude autoactivators, and find statistically significant interactions, it applies DESeq2 data analysis. To ensure proper protein folding and prevent negative interaction measurements, the workflow can be further generalized by modifying the selection method or combining it with high-throughput protease assays or protein stability and expression tests like Stable-seq. In the future, MP3-seq’s larger scale and more efficient process will hasten the adoption of HT-Y2H techniques. These advantages will make use easier in applications that span from high throughput interaction characterization of human protein variations to developing predictive models of PPIs for generative protein design.
Article Source: Reference Article | Code availability: GitHub.
Follow Us!
Learn More:




Deotima is a consulting scientific content writing intern at CBIRT. Currently she's pursuing Master's in Bioinformatics at Maulana Abul Kalam Azad University of Technology. As an emerging scientific writer, she is eager to apply her expertise in making intricate scientific concepts comprehensible to individuals from diverse backgrounds. Deotima harbors a particular passion for Structural Bioinformatics and Molecular Dynamics.





{kind=link}