
Researchers from MIT have developed a machine-learning model that can predict how two proteins bind, not in hours or minutes but just in a few seconds. Scientists may be able to use this machine-learning model to speed up the development of new drugs.
Antibodies, which are small proteins generated by the immune system, can bind to particular regions of viruses and neutralise them. One promising weapon in the fight against SARS-CoV-2, the virus that causes Covid-19, is a synthetic antibody that attaches to the virus’ spike proteins and prevents it from entering a human cell.
To create a successful synthetic antibody, scientists must first figure out how that attachment will take place. Because proteins have lumpy 3D structures with numerous folds, they may cling together in millions of different ways. Therefore, selecting the optimal protein complex from the almost infinite alternatives is exceedingly time-consuming.
Researchers from MIT developed a machine-learning model that can anticipate the complex formed when two proteins join together to speed up the process. Their method is between 80 and 500 times quicker than current software methods, and it frequently predicts protein structures closer to experimentally observed structures.
This method might aid scientists in better understanding various biological processes involving protein interactions, such as DNA replication and repair. It might also help to speed up the development of new drugs.
“Deep learning is very good at capturing interactions between different proteins that are otherwise difficult for chemists or biologists to write experimentally. Some of these interactions are very complicated, and people haven’t found good ways to express them. This deep-learning model can learn these types of interactions from data,” expressed Octavian-Eugen Ganea, the paper’s co-lead author.
Protein binding
Equidock, the model built by the researchers, focuses on rigid-body docking, which happens when two proteins join in 3D space by rotating or translating, but their forms do not compress or bend.
The model input is the 3D structures of two proteins and it turns them into 3D graphs that the neural network can process. Proteins are built up of chains of amino acids, each of which is represented in the network by a node.
Geometric information was added to the model so that it knows how things change when rotated or moved in 3D space. The model also includes mathematical information to ensure that proteins always connect in the same way, regardless of their location in 3D space. This is how proteins interact with one other in the human body.
The machine-learning algorithm uses this knowledge to identify binding-pocket locations or atoms of the two proteins most likely to interact and create chemical reactions. The points are then used to join the two proteins into a complex.
“If we can understand from the proteins which individual parts are likely to be these binding pocket points, then that will capture all the information we need to place the two proteins together. Assuming we can find these two sets of points, then we can just find out how to rotate and translate the proteins so one set matches the other set,” Ganea explains.
Overcoming the shortage of training data was one of the most difficult aspects of developing this model. Because there is so little experimental 3D data for proteins, Ganea emphasises the need of incorporating geometric information into Equidock. The model could pick up false correlations in the dataset if such geometric limitations aren’t in place.
Comparison with other baseline models
The researchers tested the model against four software approaches after being trained. After only one to five seconds, Equidock can anticipate the final protein complex. All of the baselines took a long time, ranging from ten minutes to an hour or more.
Equidock was typically equivalent to the baselines in quality metrics, which quantify how well the predicted protein complex matches the real protein complex, but it occasionally underperformed them.
“We are still lagging behind one of the baselines. Our method can still be improved, and it can still be useful. It could be used in a very large virtual screening where we want to understand how thousands of proteins can interact and form complexes. Our method could be used to generate an initial set of candidates very fast, and then these could be fine-tuned with some of the more accurate, but slower, traditional methods,” says Ganea.
The team intends to put precise atomic interactions into Equidock so that it can generate more accurate predictions, in addition to utilising this technique with standard models. For example, atoms in proteins can occasionally bind together through hydrophobic interactions, which include water molecules.
According to Ganea, the method might be used to generate tiny, drug-like compounds. Because these molecules bind to protein surfaces in specific ways, quickly figuring out how they do so could help speed up drug development.
Equidock will be improved in the future to anticipate flexible protein docking. The largest problem is a shortage of training data; therefore, Ganea and his colleagues are striving to create synthetic data that they can use to enhance the model.
Story Source: Ganea, O. E., Huang, X., Bunne, C., Bian, Y., Barzilay, R., Jaakkola, T., & Krause, A. (2021). Independent SE (3)-Equivariant Models for End-to-End Rigid Protein Docking. arXiv preprint arXiv:2111.07786.
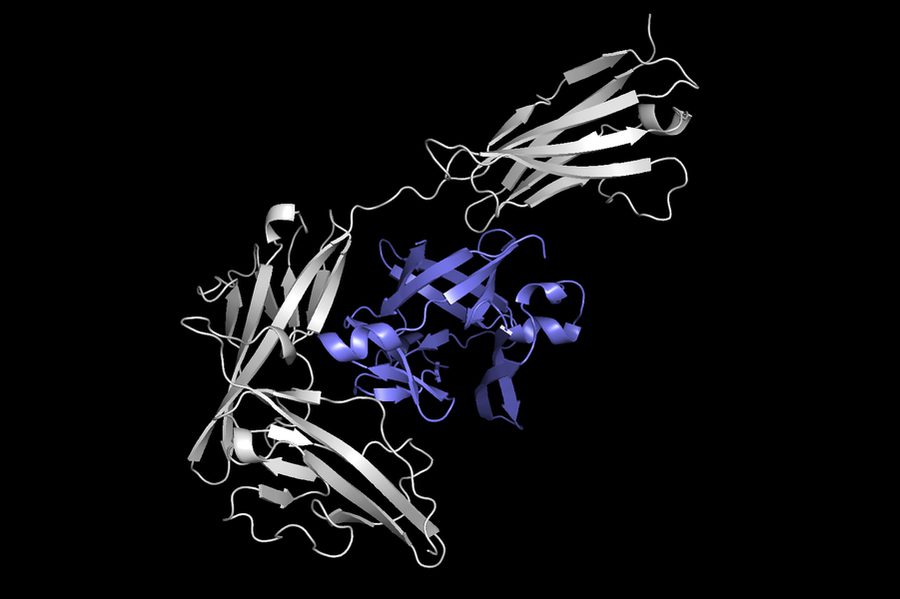
Background Image: This image shows one protein (in gray) docking with another protein (in purple) to form a protein complex. Equidock, the machine learning system the researchers developed, can directly predict a protein complex like this in a matter of seconds. Background Image Source:https://news.mit.edu/2022/ai-predicts-protein-docking-0201
Dr. Tamanna Anwar is a Scientist and Co-founder of the Centre of Bioinformatics Research and Technology (CBIRT). She is a passionate bioinformatics scientist and a visionary entrepreneur. Dr. Tamanna has worked as a Young Scientist at Jawaharlal Nehru University, New Delhi. She has also worked as a Postdoctoral Fellow at the University of Saskatchewan, Canada. She has several scientific research publications in high-impact research journals. Her latest endeavor is the development of a platform that acts as a one-stop solution for all bioinformatics related information as well as developing a bioinformatics news portal to report cutting-edge bioinformatics breakthroughs.





{kind=link}