
In de novo enzyme design, the functional groups surrounding reaction transition states are described in the optimal active site. There are drawbacks to the generative AI technique RFdiffusion, which has been used to build highly active enzymes. The active site’s geometry can only be defined at the residue level, necessitating the construction of side chain rotamers back from the functional group in order to list the locations of the residue backbone. It is also necessary to predetermine the position of catalytic residues along the sequence, which restricts the range of solutions that can be sampled. Here, researchers from the University of Washington provide a novel deep learning technique called RoseTTAFold diffusion 2 (RFdiffusion2) that addresses both issues and makes it possible to build enzymes using sequence-agnostic functional group location descriptions without the need for inverse rotamer creation. When researchers test RFdiffusion2 on an in silico enzyme design benchmark with 41 different active sites, they discover that it can effectively construct protein scaffolds for all 41 sites, as opposed to 16/41 using previous cutting-edge deep learning techniques.
Introduction
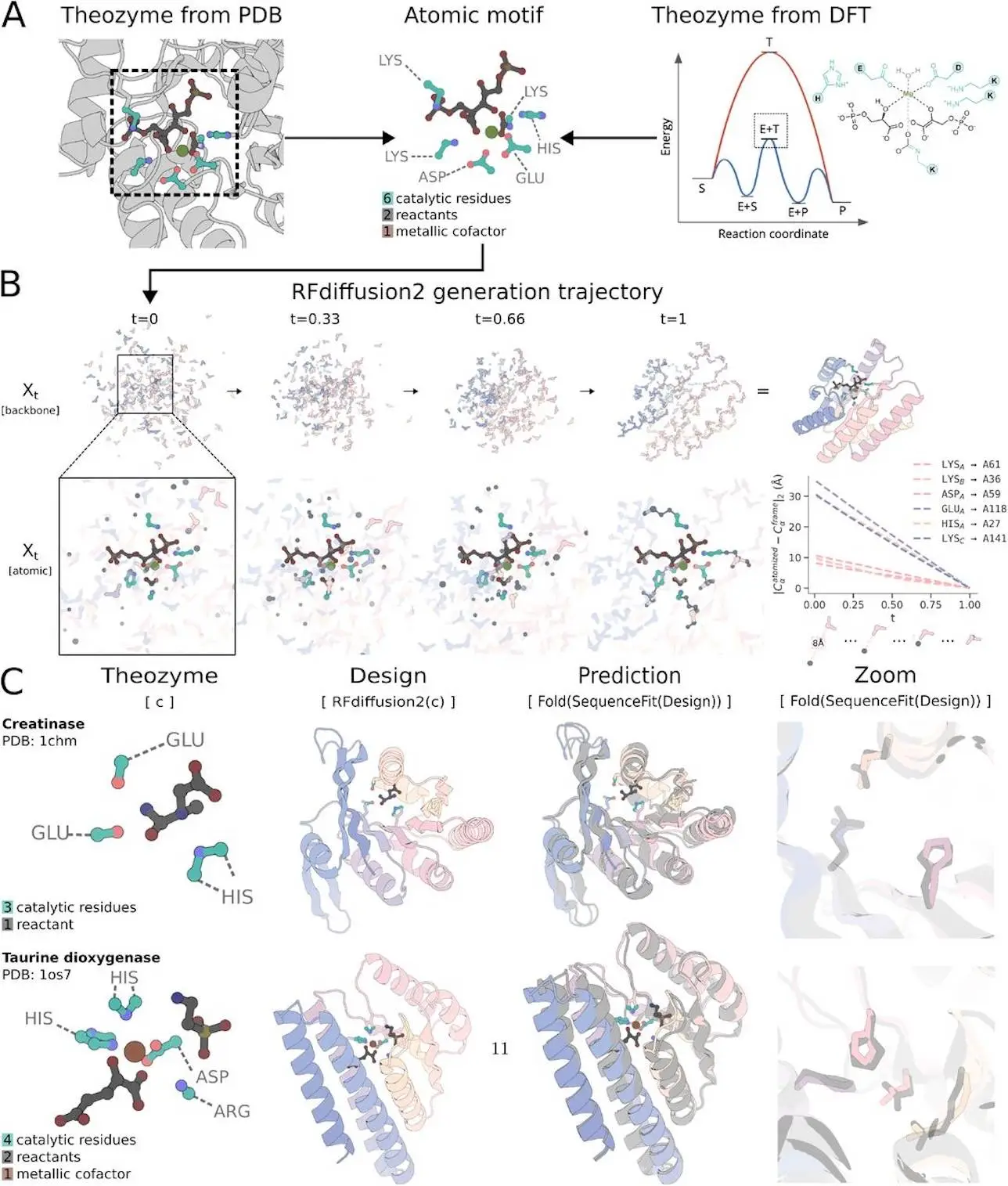
The creation of enzymes that catalyze new processes is the main obstacle in the de novo protein design process. The procedure starts with a thorough explanation of the active site structure and composition that is expected to catalyze the reaction—a theozyme. Creating protein scaffolding that can hold these theozymes is the work of de novo enzyme design. RosettaMatch and other pre-deep learning techniques looked for potential catalytic residue locations by searching through native or artificial scaffolds. However, many substructure scaffolding jobs no longer require scaffold libraries because of developments in deep learning using diffusion models. Instead, motif scaffolding allows for directly sampling various proteins that contain the desired substructure. However, the capacity of these approaches to scaffold motifs represented at the backbone level is limited since they only work with proteins represented as amino acid residues at the backbone level.
The constraints of current methods for describing active sites at the atom level are difficult to overcome by listing potential catalytic residue conformations and sequence indices. Nevertheless, this method lacks computational efficiency and is not scalable to more intricate active sites. The ability to scaffold complex theozymes characterized at the atom level would have broad uses in enzyme design and other fields. On increasingly difficult enzyme active site scaffolding problems, a generative model that can choose the conformations and sequence indices of catalytic residues could greatly enhance performance. A generative model called RFdiffusion2 builds on RosettaFold diffusion All-Atom (RFdiffusionAA) to produce structures that are dependent on descriptions of minimal active sites.
RFdiffusion2 Overview: A Deep Generative Model for Atom-Level Enzyme Design
The distribution of side chain postures is modeled by the RFdiffusion2 model using the RosettaFold All-Atom neural network architecture. An atomized residue is any residue that may be expressed as a frame or heavy atom coordinates. When atomic substructures, or atomic motifs, are included, the network learns to simulate the distribution of proteins. The coordinates of specific protein atoms, such as side chain functional groups in a theozyme, can be used to condition the model at inference time. This method removes the necessity for inverse rotamer sampling by enabling the model to infer a suitable rotamer and scaffold.
Researchers train RFdiffusion2 using flow matching, a more straightforward diffusion model framework that has been empirically demonstrated to have higher training and generation efficiency in other fields. In short, a neural network is trained to denoise by predicting the original, uncorrupted example after a training example is interpolated towards a noise sample. By repeatedly denoising a sample, a sample is taken from the noise prior; the model can sample from the data distribution if it has been taught to denoise with adequate precision. Researchers have included backbone frames and atoms, which are components of SE(3) and R3, respectively, in our description of the data distribution. While the formulation in FrameFlow for SE(3) uses Riemannian flow matching and eliminates approximations for rotational losses inherent in the RFdiffusion, flow matching on R3 adheres to its original derivation using Gaussian probability routes. These enhancements allowed RFdiffusion2 to train steadily using randomly initialized neural network weights without the need for self-conditioning or auxiliary losses. An essential first step in removing restrictions on the neural network architecture and opening up new generative modeling tasks is decoupling RFdiffusion2 from structure prediction.
Benchmark Performance of RFdiffusion2
RFdiffusion2 provides two conditioning features for de novo enzyme design: partial ligand specification and RASA specification for each ligand atom. As a result, users may precisely regulate how deep each reactant or cofactor is inside the protein. RFdiffusion2 learns to respect atomwise conditions at inference time by supplying the RASA of each ligand atom 50% of the time during training. Users can also create “partial ligands” by providing only known ligand atoms, with RFdiffusion2 inferring the remaining ligand conformer. As a result, RFdiffusion2 may sample physically realistic conformers and respect partial ligands without requiring an external tool to resolve the ligand conformer beforehand.
While RFdiffusion only finds solutions for 16 out of 41 benchmark cases, RFdiffusion2 provides solutions for all 41 cases. Researchers discover that in 40/41 situations, RFdiffusion2 performs noticeably better than RFdiffusion, establishing a new benchmark for theozyme scaffolding. Researchers discovered that the complexity of the motif, which researchers measured using the number of “residue islands,” or continuous lengths of catalytic residues in the original PDB structure, correlates with the difficulty of a benchmark example. As determined by FoldSeek and TM-score, the successful designs from RFdiffusion2 differ significantly from any protein in the training set. RFdiffusion2 may identify whole new scaffolds that include these motifs, even if the motif examples in AME are taken from the PDB.
RFdiffusion2 can scaffold theozymes based on our in silico success metrics. Researchers evaluated the model experimentally to see if it could produce functional enzymes from theozymes. When the ideal catalytic geometry is known, as shown by scaffolding theozymes from native enzymes, the experimental validation of designs from RFdiffusion2 shows that it can produce functional enzymes when screening fewer than 96 designs. In cases where the theozyme was generated by DFT, functional enzymes can also be created when starting from just the reaction mechanism. Every reaction’s most active design differs physically from every other structure in the PDB.
Conclusion
A novel approach to enzyme design called RFdiffusion2 does away with the need for expert intuition and performs better on in silico benchmarks than earlier approaches. It removes the necessity for side chain rotamers or pre-specifying sequence indices by directly scaffolding optimal active sites at the atom level. For a variety of atom-level active site descriptions, the AME benchmark demonstrates that RFdiffusion2 outperforms RFdiffusion by a large margin. The ability of RFdiffusion2 to design enzymes across numerous reactions indicates that it may be able to design enzymes across more reactions with greater success rates than the previous state-of-the-art. However, enhancements include co-designing protein sequences and side chains beyond the active site, automating the design process, and broadening the definition of the theozyme to include more interactions for high activity.
Article Source: Reference Paper
Disclaimer:
The research discussed in this article was conducted and published by the authors of the referenced paper. CBIRT has no involvement in the research itself. This article is intended solely to raise awareness about recent developments and does not claim authorship or endorsement of the research.
Important Note: bioRxiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Follow Us!
Learn More:




Deotima is a consulting scientific content writing intern at CBIRT. Currently she's pursuing Master's in Bioinformatics at Maulana Abul Kalam Azad University of Technology. As an emerging scientific writer, she is eager to apply her expertise in making intricate scientific concepts comprehensible to individuals from diverse backgrounds. Deotima harbors a particular passion for Structural Bioinformatics and Molecular Dynamics.









{kind=link}