
Researchers at Tsinghua University have introduced DrugCLIP, a contrastive learning framework that enables ultrafast and accurate virtual screening at a genome-wide scale. Published in Science, it is validated against 5-HT2AR, TRIP12, and NET, and it enables virtual screening at the genome scale, up to seven orders of magnitude faster than docking methods.
Why is there a need for Genome-Level Virtual Screening
Drug discovery is all about finding a chemical molecule that binds effectively to a target. Earlier researchers usually picked one protein and spend years finding molecules that bind to it using traditional approaches like High-throughput screening, molecular docking, etc. However, despite the progress in these methods, only 10% of disease-associated proteins currently have medicinal drugs, as many proteins are unexplored, dynamic, and the older approaches are either computationally expensive, limited to only known ligands, or cannot handle the huge scale, diversity, and complexity of millions of compounds.
This is where genome-scale virtual screening (GSVS) comes into the picture. It is the systematic computational evaluation of billions of molecules against thousands of proteins across the entire human genome. It expands drug discovery from a handful of well-studied drugs to the full proteome, including proteins that were difficult to study, such as Intrinsically Disordered Proteins (IDPs) or fusion oncoproteins.
Earlier GSVS approaches like docking-based genome-wide scans and ligand-based similarity searchers were too slow, limited, or inaccurate. Therefore, Jia’s team developed DrugCLIP, a one-of-a-kind AI framework based on contrastive learning.
DrugCLIP’s Architecture: An Introduction to Contrastive Learning
DrugCLIP is trained on both synthetic large-scale data and real experimental complexes. This hybrid training improves generalization to unseen proteins, too. The inputs taken by the model are protein pockets and small molecules, which are extracted from AlphaFold2 and graphs, respectively. Protein encoders in the model learn structural and physicochemical features of binding pockets. At the same time, the molecule encoder learns chemical features. Both encoders map inputs into embedding vectors. Contrastive learning aligns proteins and molecules.
Contrastive learning is a machine learning technique where the model learns by comparing pairs of data points in a shared latent space (a mathematical representation where similar protein molecule pairs are close together). The core idea is to bring binding protein and molecule pairs (positive pairs) together, while the ones that don’t bind (negative pairs) are pushed farther apart in space.
Once embeddings are learned, screening becomes a search problem. For a given pocket embedding, the model retrieves nearest molecule embeddings from a massive library just like how search engines retrieve documents. This makes screening 10 million times faster.
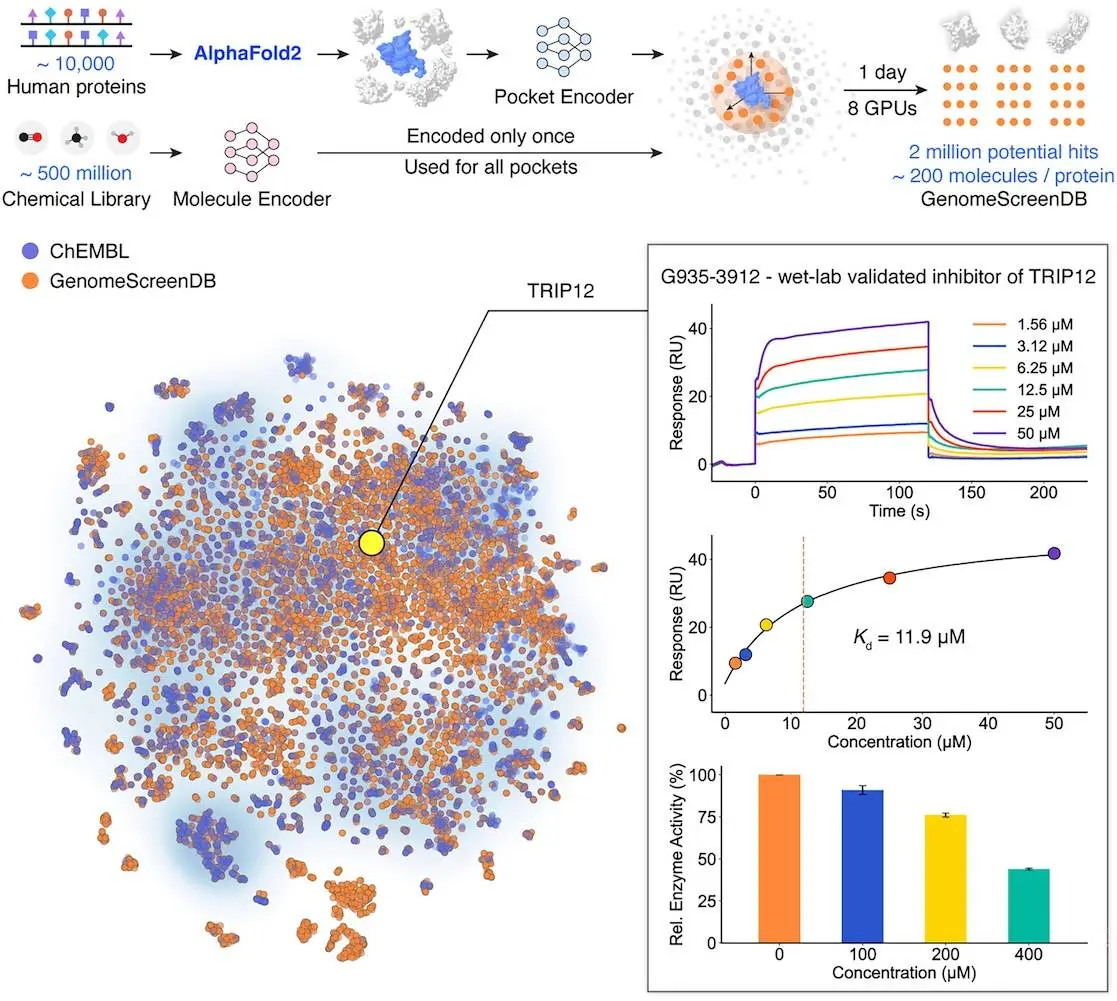
To make the model even more accurate, the model integrates the GenPack module, a generative pocket refinement tool that improves AlphaFold2’s predicted pockets. This has been proved by the case study of Thyroid hormone receptor interactor 12 (TRIP12), a less explored protein with no reported holo structure or ligand. DrugCLIP successfully identified small molecule inhibitors for TRIP12 with a hit rate of 17.5%.
Wet Lab Validation: Why DrugCLIP is Transformative
DrugCLIP was validated against wet lab experiments, keeping in mind that computational predictions are only useful if they hold up in real experiments.
The model was applied to an open-source database of ~10,000 human proteins. It screened these proteins against 500 million chemical compounds like ZINC and Enamine REAL in under 24 hours using only 8 GPUs. It still managed to yield >2 million candidate potent agonists and inhibitors for several proteins. Some of these hits were validated even when the protein structures came only from AlphaFold2 prediction, not experimentally solved. This shows DrugCLIP can work reliably even for proteins that have never been studied in detail.
Traditional methods would never be able to handle the scale and still be fast enough to be applied to drug discovery, even after being computationally lightweight.
Experimental Validation Results
DrugCLIP was tested on the chosen targets Serotonin 2A receptor (5HTAR) and Norepinephrine transporter (NET), both heavily implicated in psychiatric disorders. The two molecules predicted by DrugCLIP were experimentally confirmed as activators for 5HT2AR with a median effective concentration (EC50) value of <100nM, meaning they both were highly potent, comparable to or better than existing drugs. While the two molecules predicted as inhibitors for NET were validated using cryo-electron microscopy (cryo-EM), confirming that the molecules bound to NET as predicted.
Key Takeaways
DrugCLIP is not just faster but more accurate than existing methods. It’s a transformative framework enabling trillion-scale, proteome-wide drug discovery. Its validation across benchmarks and experiments makes it a gemstone for next-generation therapeutics, especially for targets that were previously overlooked.
Article Source: bioRxiv | Abstract | The reference pipeline is available at GitHub.
Disclaimer:
The research discussed in this article was conducted and published by the authors of the referenced paper. CBIRT has no involvement in the research itself. This article is intended solely to raise awareness about recent developments and does not claim authorship or endorsement of the research.
Follow Us!
Learn More:




Saniya is a graduating Chemistry student at Amity University Mumbai with a strong interest in computational chemistry, cheminformatics, and AI/ML applications in healthcare. She aspires to pursue a career as a researcher, computational chemist, or AI/ML engineer. Through her writing, she aims to make complex scientific concepts accessible to a broad audience and support informed decision-making in healthcare.











{kind=link}