
Cellarity, Inc., and NVIDIA have unveiled MOLRL (Molecule Optimization with Latent Reinforcement Learning), a groundbreaking framework for molecular optimization. This approach leverages the latent spaces of pre-trained generative models and employs Proximal Policy Optimization (PPO), a cutting-edge reinforcement learning (RL) algorithm. PPO is particularly effective in high-dimensional, continuous domains, enabling a balanced trade-off between exploration and exploitation while maintaining stability in optimization. The framework’s versatility allows it to work across various generative model architectures, with the latent space’s structure playing a pivotal role in determining its performance.
By utilizing computational techniques to design compounds with specific physicochemical properties or biological activities, MOLRL significantly advances the drug discovery process. The latent space of a generative model facilitates molecule creation without requiring explicit chemical rules, making it a powerful tool for optimizing molecular characteristics and incorporating predetermined substructures. This capability is critical for real-world drug development scenarios. The efficacy of the MOLRL approach is demonstrated through its integration with latent spaces of different autoencoder models, showcasing its potential to deliver state-of-the-art results in molecular design while remaining architecture-agnostic. This innovation marks a significant step forward in computational chemistry and biotechnology.
Introduction
In drug discovery, optimization is essential since a chemical molecule needs to have several characteristics and biological activity to be considered a clinical candidate. A lengthy and iterative approach is used to structurally modify a hit that has been found to exhibit therapeutic efficacy to overcome vulnerabilities such as insufficient solubility and activity. Based on reaction-based library enumeration or intuition, medicinal chemists create analogs by transforming the starting molecule. Creating targeted molecules using computational techniques can aid in the effective exploration of the chemical space and suggest structures that have not yet been investigated.
Machine learning and generative deep learning models have been extensively investigated in chemistry and drug development for the creation of de novo molecules. The two main types of targeted molecule generation and optimization techniques currently in use are those that work directly on the molecular structure to find structural changes that enhance the desired property and those that work in the latent space of a generative model.
Looking into Previous Models
Reinforcement Learning (RL) is frequently used in the former methods to optimize discrete space. These techniques can be used directly on the molecular graph to find structural changes—such as adding or removing atoms or bonds—that will enhance the desired attribute. The problem here is that structural changes could lead to erroneous molecule structures by breaking chemical laws. Hence, it is thought important to account for chemical knowledge using chemical rules or heuristics. Working at the substructure level and assembling molecular fragments to create a molecular graph is an alternative strategy. Lastly, sequence-based strategies have also been investigated using RL techniques to manage the creation of SMILES. These methods are based on language models, which have the benefit of being extensively trained on chemical datasets, allowing them to accurately capture the validity and language of chemicals.
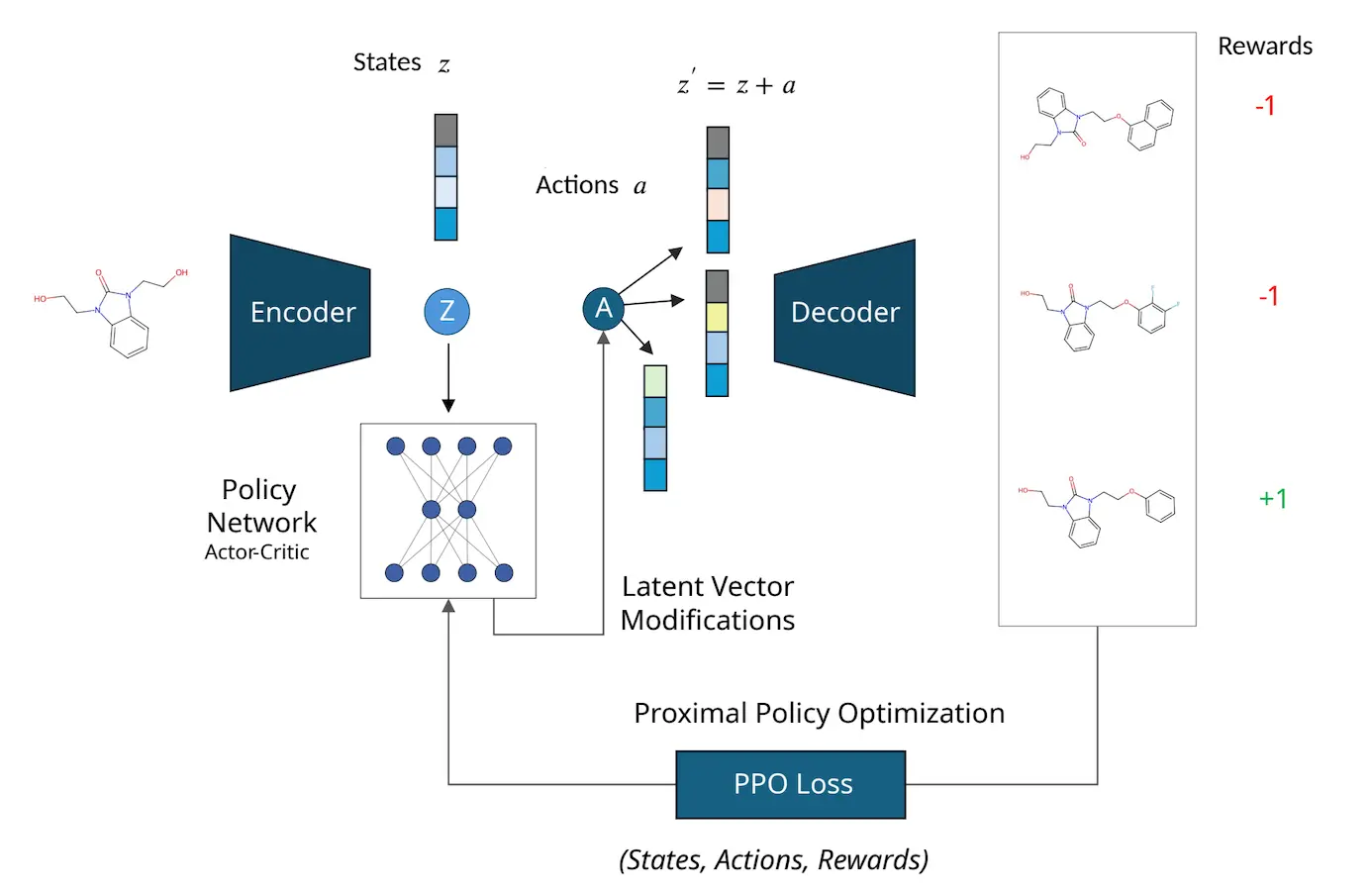
Understanding MOLRL
The authors present MOLRL (Molecular Optimization with Latent Reinforcement Learning), a novel framework for molecular optimization that uses RL with potent pre-trained latent space generative models. The sophisticated policy gradient RL method, Proximal Policy Optimization (PPO), is used by MOLRL to optimize the latent space of a pre-trained generative model. Searching difficult settings, such as chemical latent spaces, requires a trust region, which PPO can maintain while striking a healthy balance between exploration and exploitation. Although the design of the generative model is irrelevant to the suggested framework, the latent space’s properties can significantly affect how well the optimization technique performs.
The characteristics of the latent space aid the optimization process in the latent space. MOLRL, a latent space-based approach, is assessed using both state-of-the-art techniques and benchmarks from the literature. The efficacy of the approach in scaffold-constrained molecule optimization is illustrated by its demonstration for both single-property constraint and multi-objective optimization challenges.
Key Contributions of the Paper
- In the latent space of an autoencoder architecture that has already been trained, researchers present a novel approach for targeted molecule creation. For continuous space optimization, the approach blends cutting-edge RL algorithms with potent generative models that have already been trained on enormous chemical datasets.
- Researchers go over the late space characteristics that are essential to the optimization framework that has been presented. Researchers also go over the training changes researchers made to a variational autoencoder in order to enhance those characteristics.
- By using the method on drug discovery-related activities, comparing it to state-of-the-art techniques, and using popular benchmarks, researchers show how successful the focused molecule generation method is.
Conclusion
MOLRL is a new method for creating tailored molecules that combines Reinforcement Learning with strong generative models. In molecular optimization, a continuous representation of molecules appropriate for optimization tasks is provided by the latent space of an autoencoder generative model trained on molecules. Comparing MOLRL to well-established techniques demonstrates competitive or better performance on a range of tasks for multi-parameter optimization and targeted molecule creation. A greater chemical space is covered by denser latent spaces as a result of ongoing improvements in generative models. For effective navigation of these spaces to find novel compounds with desirable features, the growing use of these models in drug discovery is essential. However, the synthesizability of the molecules produced is a major barrier to the practical use of models based on generative machine learning. The efficiency of optimization is another problem for real-world applications, particularly when it comes to supporting computationally intensive reward functions like molecular docking, and the efficacy of methods for assessing the synthesizability of created compounds is still missing.
Article Source: Reference Paper
Disclaimer:
The research discussed in this article was conducted and published by the authors of the referenced paper. CBIRT has no involvement in the research itself. This article is intended solely to raise awareness about recent developments and does not claim authorship or endorsement of the research.
Important Note: ChemRxiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Follow Us!
Learn More:




Deotima is a consulting scientific content writing intern at CBIRT. Currently she's pursuing Master's in Bioinformatics at Maulana Abul Kalam Azad University of Technology. As an emerging scientific writer, she is eager to apply her expertise in making intricate scientific concepts comprehensible to individuals from diverse backgrounds. Deotima harbors a particular passion for Structural Bioinformatics and Molecular Dynamics.











{kind=link}