
The availability of biological “big data” and the development of high-throughput sequencing technology have sped up the identification of new protein sequences, making it difficult to keep up with their functional annotation. Techniques like Sequence Similarity Networks (SSNs) have been used to visually arrange proteins for quicker identification in order to overcome this annotation difficulty. Researchers from the University of California San Diego, Instituto Politécnico Nacional, and Universidad Autónoma de Baja California present the Protein Language Visualizer (PLVis) that brings together pre-trained PLM embeddings, dimensionality reduction algorithms, and clustering techniques to rapidly evaluate proteins based on their neighbors. It is possible to enhance these interactive representations with structural and biological annotations. Multi-organism whole proteome comparisons show the pipeline’s worth, and the PLVis Colab Notebook is made available to the larger scientific community.
Introduction
Although protein sequence discovery has been transformed by high-throughput sequencing technology, many unreviewed entries in the database still present annotation issues. Even though UniProtKB automatically annotates unreviewed entries, more than 30% of genes that encode proteins are still unannotated. Sequence Similarity Networks (SSNs), a visual and interactive analytic technique, have become widely used to address these issues. Protein interactions are visually represented by SSNs, which join nodes when their BLAST-based pairwise alignment surpasses a threshold of similarity that the user specifies.
The EFI-EST web server and other tools have made SSNs available to investigate protein sequence-function connections. Profile Hidden Markov Models (HMMs), which identify sequence domains and cluster protein sequences into families by identifying statistical patterns in the sequences, are more sophisticated statistical techniques in protein research that SSNs complement. Multiple protein HMM-based visualizations are infrequently used and are frequently indirect.
The conceptual successors of HMMs are Protein Language Models (PLMs), which are also derived from NLP. Both individual amino acids (tokens) and entire protein sequences can be represented as high-dimensional vectors (embeddings) by them; they typically employ a transformer neural network design. These PLM embeddings are useful tools for protein design and synthesis, as well as downstream prediction and classification tasks like structure prediction.
Understanding Protein Language Visualizer (PLVis)
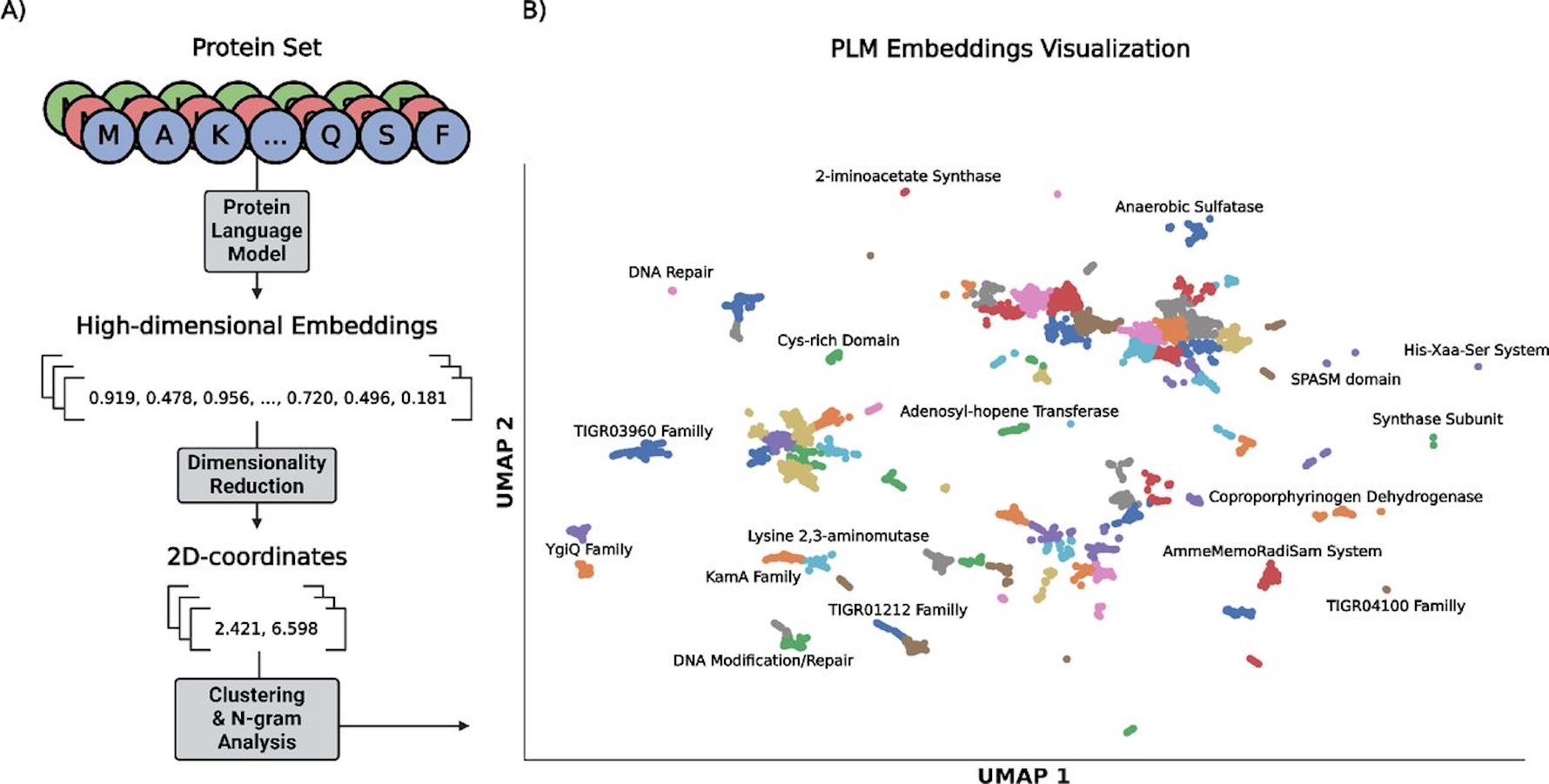
An effective and user-friendly substitute for the visual display of protein data derived from PLM embeddings is the PLVis pipeline. By successfully grouping proteins based on their family classifications, these visualizations improve protein functional annotation when combined with SSNs. PLVis projections, for example, can be used to quickly classify proteins into different subfamilies by researchers looking into particular protein families and attempting to confirm the functionality of poorly annotated proteins. Especially when minimally annotated proteins are discovered close to well-characterized proteins, this clustering makes it easier to identify good candidates for experimental validation.
PLVis Colab generating Proteome Comparison
Together with the help of the intuitive PLVis Colab Notebook, users may upload data frames and insert files of matching proteomes to construct exploratory data analysis visualizations. The software analyzes average silhouette ratings across various cluster numbers using k-means clustering to find the ideal number of clusters. Depending on the graph, users can apply the ideal number of clusters that is automatically determined or select their number. Following the execution of each cell, a CSV file with the coordinates of every protein in the data frame is produced.
Users can choose the final visualization’s color by modifying it according to column-specific data. The projection can also be filtered by choosing particular head-to-head comparisons from the chosen column. The Colab provides a fast structure comparison analysis following the main results, which retrieves and displays the available AlphaFold-generated structures for a predetermined set of proteins. Researchers who want to comprehend intricate protein interactions and patterns in their datasets will find the PLVis Colab to be a very useful tool.
Features of PLVis
Protein Language Visualizer shows great usability in more general comparison research. Comparative investigations of entire proteomes across species show that PLVis projections show the greatest biological utility. The resulting protein clustering patterns provide important biological information, such as conserved patterns within taxonomic genera or the absence of protein families peculiar to a species. Visualization is beneficial for examining particular biological relationships, such as host-pathogen interactions, where clusters of proteins from both organisms may be linked to disease. Protein clusters of this kind offer possible molecular markers linked to disease processes.
Conclusion
PLM embeddings can be easily replaced with the Protein Language Visualizer pipeline, which shows protein data visually. When used in conjunction with SSNs, it effectively classifies proteins according to familial ties, enhancing functional annotation. The ability of PLVis projections to rapidly categorize proteins into distinct subfamilies allows researchers to verify the activity of proteins with inadequate annotations. It helps to find promising candidates for experimental validation when minimally annotated proteins are present near well-characterized proteins. The PLVis Colab Notebook fills a significant void in the understanding of high-throughput biological data by providing a solid foundation for the visual examination of intricate proteomic datasets. It provides a thorough method for examining proteomic data, leading to a deeper understanding of various biological research projects. The scientific community actively seeks the tool because of its iterative development and improvement.
Article Source: Reference Paper | PLVis Colab Notebook.
Disclaimer:
The research discussed in this article was conducted and published by the authors of the referenced paper. CBIRT has no involvement in the research itself. This article is intended solely to raise awareness about recent developments and does not claim authorship or endorsement of the research.
Important Note: bioRxiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Follow Us!
Learn More:




Deotima is a consulting scientific content writing intern at CBIRT. Currently she's pursuing Master's in Bioinformatics at Maulana Abul Kalam Azad University of Technology. As an emerging scientific writer, she is eager to apply her expertise in making intricate scientific concepts comprehensible to individuals from diverse backgrounds. Deotima harbors a particular passion for Structural Bioinformatics and Molecular Dynamics.











 for faster functional annotation of protein sequences.){kind=link}