
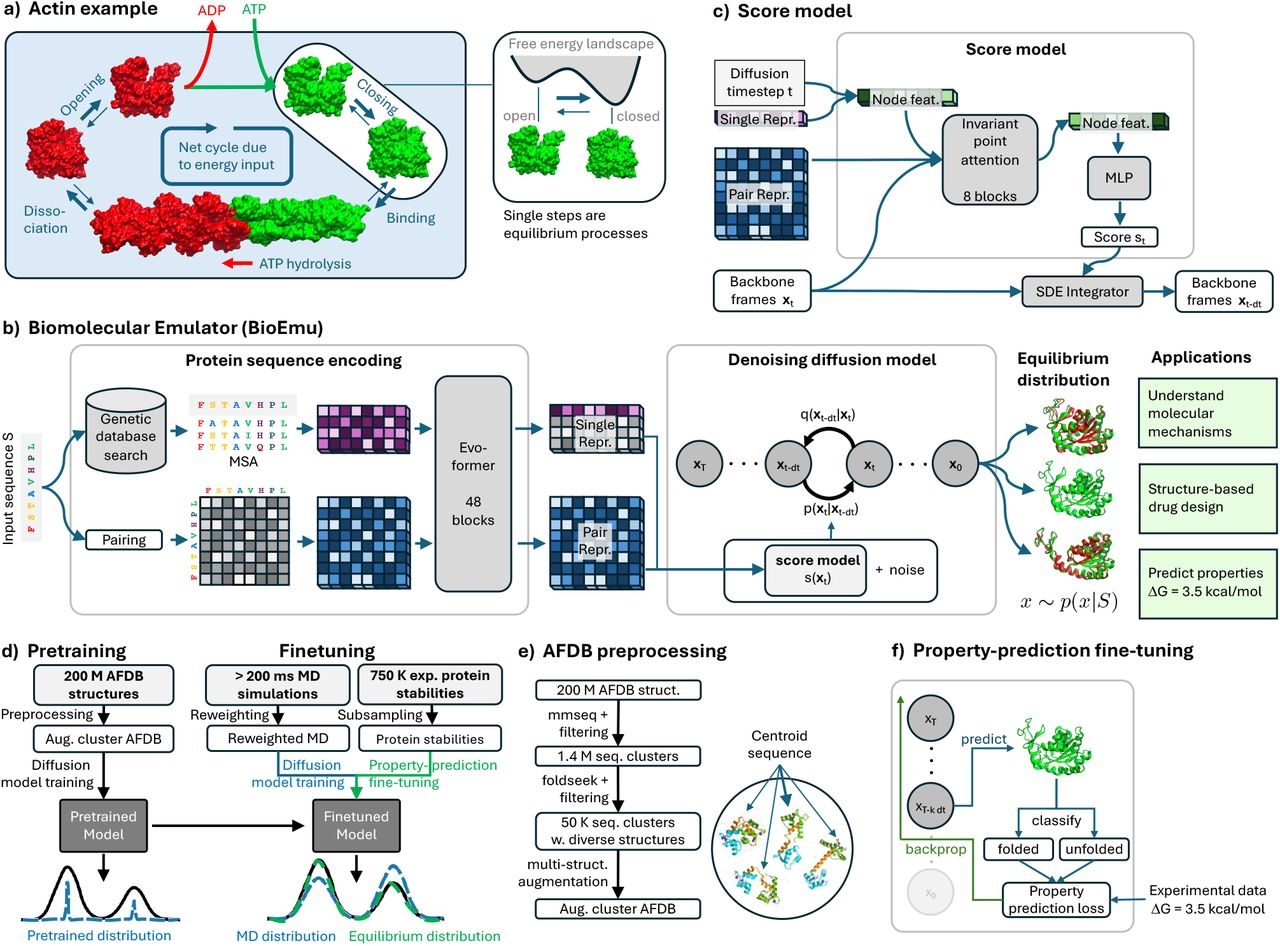
A major field of study is the scientific problem of predicting dynamical mechanisms in proteins after structural revolutions, with molecular dynamics simulations offering important information on conformational states and binding configurations. In this work, researchers from Microsoft Research create a generative deep learning system called Biomolecular Emulator (BioEmu), which can produce thousands of statistically distinct protein structure ensemble samples every hour on a single graphics processing unit. BioEmu studies protein structures using large amounts of data and innovative training techniques. Numerous conformational changes are sampled, such as domain rearrangements, cryptic pockets, and the unfolding of particular protein areas. Additionally, Protein Conformations are quantified by BioEmu with relative free energy errors of approximately 1 kcal/mol. Through the simulation of structural ensembles and thermodynamic properties, this method can effectively provide hypotheses that can be tested experimentally and offer mechanistic insights, such as the origins of fold instability.
Introduction
Proteins play a major role in drug development, enzymatic catalysis, biotechnological processes, and biomaterials. One of the biggest challenges in science and technology is comprehending how proteins function and how to control them. Sequence, structure, and function are the three main pillars of protein science. While the Protein Data Bank (PDB) has made it possible to predict 3D protein structures, next-generation sequencing has made it possible to obtain protein sequences of complete genomes at a reasonable cost. The comprehension of how proteins operate is hampered by the lack of highly accurate and high-throughput protein function technologies.
Functional descriptions, like “actin builds up muscle fibers,” are attributions made by humans based on mechanistic qualities that can be measured empirically. Understanding the conformational states of proteins—which can exist in a variety of structures—as well as the likelihood that certain conformational and binding states would occur under specific experimental conditions, is necessary for these descriptions. For instance, the molecular underpinnings of muscle growth are provided by actin, which has several conformational and binding states that are controlled by its cofactors ATP and ADP. The methods available today, however, are not scalable for examining these conformational and binding states and their probability.
Full equilibrium distributions of observables are obtained using single-molecule experiments; however, they necessitate custom molecular structures and laborious data gathering. Multiple conformational states and their probabilities can be resolved using cryo-electron microscopy; however, doing these studies is expensive. A ubiquitous technique for investigating the structure and dynamics of biomolecules at all-atom resolution is molecular dynamics (MD) simulation; yet, protein folding and association are difficult to examine due to the sampling problem and imperfect biomolecular forcefields. The possibility exists for machine-learned coarse-grained MD models to attain comparable accuracy at reduced computational expenses.
Understanding BioEmu
The biomolecular emulator, or BioEmu, is a new machine learning system that can precisely sample from the protein conformation equilibrium distribution in a few GPU hours each experiment. Together with experimental observations of protein stabilities, a sizable collection of static protein structures and copious MD simulation data are used to train the system. Tasks like predicting folded states of tiny proteins, simulating equilibrium distributions, and forecasting changes in protein shape are used to validate the system. With a free energy accuracy of less than 1 kcal/mol, the system can predict protein conformations with precision comparable to that of experiments. One advantage of BioEmu over MD forcefields is its ability to be effectively adjusted on experimental data, including folding free energies. Nevertheless, sampling the processes that result in experimental observables throughout the training phase is either laborious or impractical for observables involving complicated rare events.
Limitations
The empirical generation of distributions using MD simulation, in contrast to BioEmu, is a limitation related to statistical mechanics. BioEmu can be used for enhanced sampling simulations and reweighting since it generates distributions based on p(x) ∝ e(u(x)). However, the existing system only simulates single protein chains at a constant thermodynamic condition of 300K, which necessitates conditioning on parameters relevant to biology and experiment, such as pH and temperature. It is necessary to represent several interacting molecules to create an appropriate protein emulator.
Conclusions
Protein conformation and equilibrium probabilities can be explored by sampling the equilibrium distributions of proteins using a generative machine learning system. It has been demonstrated that the system can appropriately model the equilibrium distributions of large-scale MD simulations and forecast protein stabilities determined experimentally with an error of less than 1 kcal/mol. MD simulations and experiments that yield comprehensive structure-function relationships are much more expensive than inference, which is typically done at a cost of one GPU hour per computational experiment. To expand the emulator to additional scopes and enhance it for the current scope, however, a major barrier is the absence of training data. To learn conformational and binding state changes of large biomolecular complexes and to comprehend the subtle differences in binding affinities between binding partners, highly scalable experimental techniques that can be used as training data will be essential for accurately predicting protein function.
Article Source: Reference Paper
Disclaimer:
The research discussed in this article was conducted and published by the authors of the referenced paper. CBIRT has no involvement in the research itself. This article is intended solely to raise awareness about recent developments and does not claim authorship or endorsement of the research.
Important Note: bioRxiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Follow Us!
Learn More:




Deotima is a consulting scientific content writing intern at CBIRT. Currently she's pursuing Master's in Bioinformatics at Maulana Abul Kalam Azad University of Technology. As an emerging scientific writer, she is eager to apply her expertise in making intricate scientific concepts comprehensible to individuals from diverse backgrounds. Deotima harbors a particular passion for Structural Bioinformatics and Molecular Dynamics.











{kind=link}