
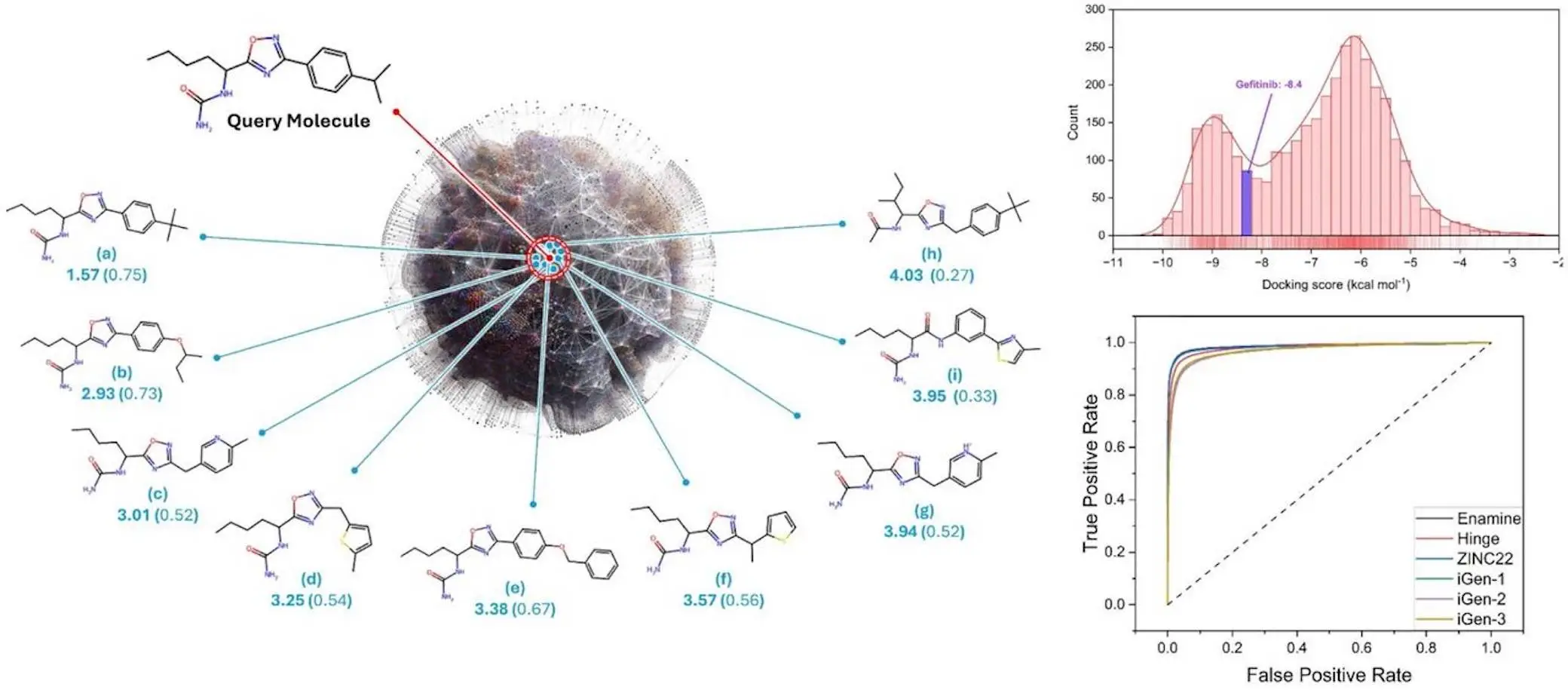
Recent years have witnessed significant advancements in the fields of artificial intelligence (AI) and machine learning (ML), catalyzing a revolution in cheminformatics and drug discovery. These advancements have enabled scientists to predict molecular properties, design novel compounds with desired characteristics, and estimate drug/bio-macromolecular interactions with unprecedented accuracy. At the heart of these breakthroughs lies the development of molecular descriptors, quantitative representations of chemical information that serve as the foundation for various predictive and generative models.
In a recent study, researchers from the Department of Chemistry and Molecular Biology at the University of Gothenburg, Sweden, introduced MolAI, a robust deep-learning model designed for data-driven molecular descriptor generation. This innovative model leverages a vast training dataset of 221 million unique compounds and employs an autoencoder neural machine translation (NMT) architecture to generate latent space representations of molecules.
Traditional vs. Data-Driven Molecular Descriptors
Traditionally, molecular descriptors were created manually, which meant that experts used to translate molecular properties into vectors that computers could read. Morgan fingerprints, later referred to as extended connectivity fingerprints or ECFP, have been one of the most popular choices for vectors due to outperforming other fingerprints used in molecular bioinformatics and virtual screening. However, ECFPs still have their disadvantages. They tend to be high dimensional and sparse and suffer from bit collisions caused by hashing, and in most cases, there is no representation learning happening when using ML models in cheminformatics and drug design as they use traditional extracted descriptors, which contradicts the core principle of representation learning. A backbone of deep learning frameworks offered for dealing with computer vision tasks is neural networks, which have shifted the paradigm toward trainable algorithms capable of learning representations of the molecules from large datasets comprised of lower-level formats like graphs or SMILES (simplified molecular input line entry specification).
MolAI: A Deep Learning Solution
MolAI utilizes the works of Mahdizadeh and Eriksson while incorporating neural machine translation (NMT), similar to Winter et al.’s work. However, MolAI has a larger training dataset, incorporates 13 molecular property regression models, uses LSTM units, and trains on TensorFlow 2.7, which enhances training and prediction speeds.
The model applied to the subset of 20 million samples was built using the Keras tuner tool, which aided in garnering the best-performing hyperparameters through prior tuning with an exhaustive search on the other components of the polynomial. The final model was selected based on the combination of hyperparameters that yielded the lowest validation loss.
Exceptional Performance and Versatility
MolAI exhibited outstanding results, attaining accuracy > 99.99% on the validation set relating input SMILES strings to their latent vector representations. Having the capability to encode and decode molecules from latent space allows for endless possibilities in drug development, including but not limited to structure-activity relationship analysis, hit optimization, de novo molecular design, and the training of sophisticated machine learning algorithms.
The efficiency of MolAI-powered molecular descriptors was further explored via additional benchmarking tests. These tests confirmed that MolAI predicts the dominating protonation state of molecules more accurately, improving ligand-based virtual screening, and predicts ADMET (Absorption, Distribution, Metabolism, Excretion, and Toxicity) properties of drug-like molecules with far greater precision.
Conclusion
The creation of MolAI epitomizes a profound increase in the use of deep learning in molecular descriptor generation and drug discovery. By optimally encoding molecules into a deep latent space, a more profound understanding of molecular properties and the generation of new compounds with specific traits is achievable. This progress could enhance the rate of discovery of new drugs and make the development processes more efficient, paving the way for innovative solutions to pressing healthcare challenges.
Article Source: Reference Paper | Data, software, and materials are available on ANYO Labs GitHub repository.
Disclaimer:
The research discussed in this article was conducted and published by the authors of the referenced paper. CBIRT has no involvement in the research itself. This article is intended solely to raise awareness about recent developments and does not claim authorship or endorsement of the research.
Important Note: bioRxiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Follow Us!
Learn More:




Anchal is a consulting scientific writing intern at CBIRT with a passion for bioinformatics and its miracles. She is pursuing an MTech in Bioinformatics from Delhi Technological University, Delhi. Through engaging prose, she invites readers to explore the captivating world of bioinformatics, showcasing its groundbreaking contributions to understanding the mysteries of life. Besides science, she enjoys reading and painting.











{kind=link}