
Microscopic imaging and high-throughput sequencing methods show that chromatin structures differ greatly between cells, although characterization is still difficult because of the labor-intensive nature of the investigations. Researchers from the Massachusetts Institute of Technology present ChromoGen, a generative model that effectively predicts three-dimensional, single-cell chromatin conformations de novo with both region and cell type specificity. It is built on cutting-edge artificial intelligence techniques and was created to address these issues. These produced conformations faithfully replicate single-cell and population-level experimental findings. Additionally, ChromoGen provides access to chromatin structures in a wide variety of cell types by effectively transferring to cell types that were not included in the training set using only DNA sequences and publicly available DNase-seq data. The cost of computation for these accomplishments is very inexpensive. As a result, ChromoGen makes it possible to systematically examine the organization of single-cell chromatin, its heterogeneity, and its connection to sequencing data while maintaining the economy.
Introduction
Chromatin structures are essential for gene regulation because they determine the patterns of gene expression and regulatory systems. By demonstrating how chromatin patterns support the spatial environment surrounding genes and help recruit the proper molecules at the right moment, advances in molecular biology techniques have shed light on genome conformations. A significant degree of variation in chromatin patterns across cells in the same population has been revealed by high-throughput sequencing and imaging technologies, which have enhanced our capacity to examine genome organization in individual cells. Nevertheless, current approaches are frequently time-consuming and labor-intensive, which prevents in-depth examinations of the conformational variability of chromatin in various biological situations, preventing a deeper comprehension of the stochastic nature of gene regulation and the principles underlying genome structure.
For high-resolution chromatin conformation characterization, computational techniques offer an alternate strategy. Current methods use population genome-wide chromatin conformation capture (Hi-C) data to extract single-cell information and fit an ensemble of chromatin conformations to the average contact probabilities between genomic loci. However, because we don’t fully understand the exact chemical pathways driving chromatin folding, it’s still difficult to generalize these techniques to predict chromatin shapes de novo. Accurate population Hi-C data prediction is now possible thanks to deep learning approaches, which have opened up new pathways for directly predicting single-cell chromatin conformations from sequencing data. However, because of the intrinsic stochastic nature of mapping sequence characteristics to individual 3D structures due to cell-to-cell variation, these methods have not been used to predict 3D chromatin conformations.
Looking into the ChromoGen Model
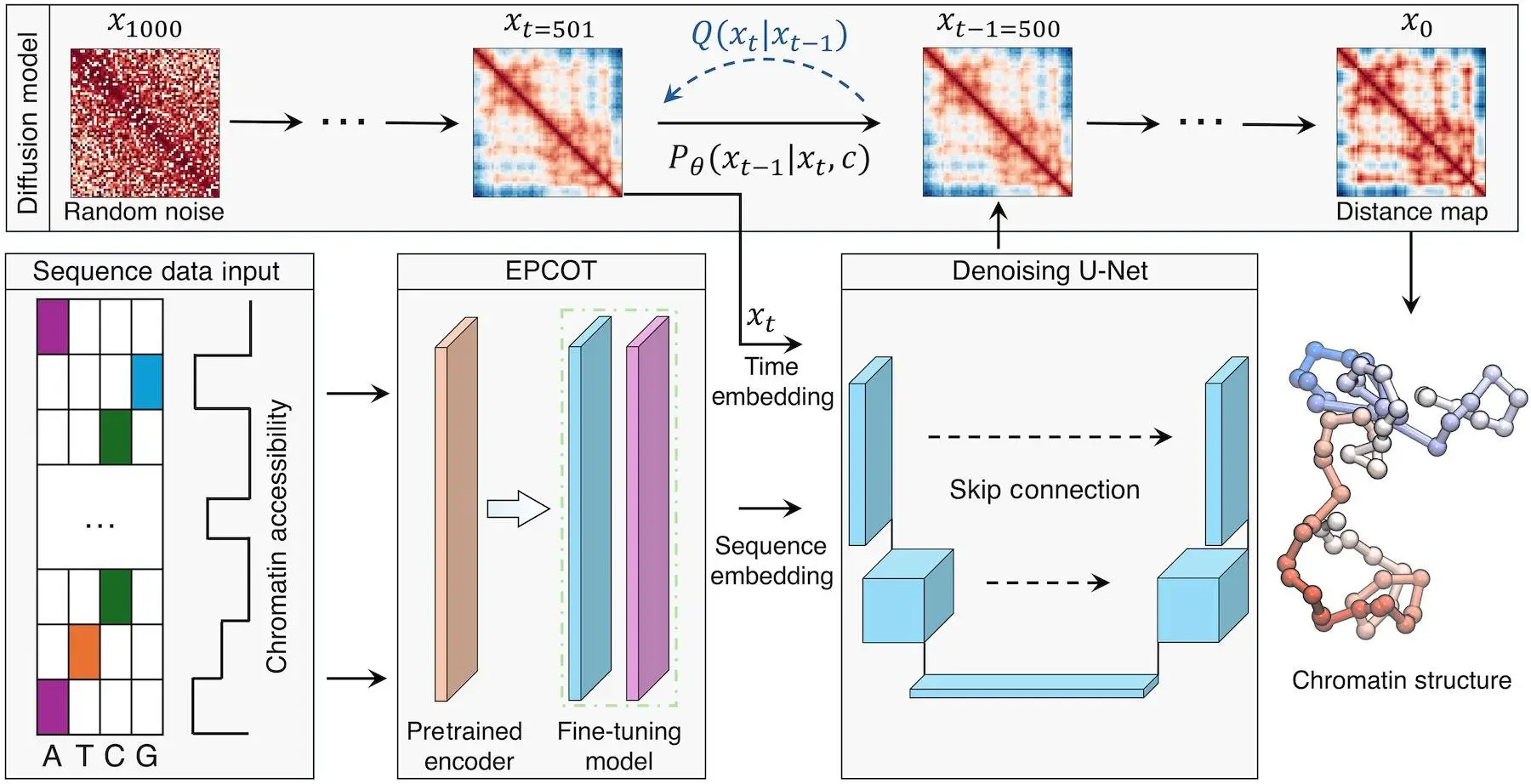
ChromoGen is a diffusion model that captures the variability of single-cell chromatin structures and makes de novo predictions about them. This AI method is quite good at predicting the three-dimensional coordinates of protein molecules and ligands, as well as text-to-image applications. After training, diffusion models effectively produce fresh samples that closely resemble the training data’s statistical distribution, offering physically accurate substitutes for the original data. Conditioned on external data such as DNA sequence and chromatin accessibility information, ChromoGen may generate 3D conformations stochastically that represent heterogeneity in single-cell datasets. This makes it possible for diffusion models to use sequencing data alone to produce chromatin conformations specific to a region and cell type.
ChromoGen accurately reproduces the conformational distribution of chromatin
ChromoGen is a tool for creating chromatin structures, focusing on their quality. The tool can predict valid conformational ensembles for genomic regions, but it must also capture and distribute the full range of generic topological features in chromatin. This approach offers valuable insights into the topological complexities of chromatin, a fundamental biological polymer.
ChromoGen correctly forecasts conformational ensembles for particular areas
ChromoGen is a chromatin-based technology designed to capture structural features on chromosomes 1–22 that are biologically significant, with the exception of chromosome X. ChromoGen proved to be transferable and capable of making de novo predictions on chromosome X, even though data from this gene was not included in genome-wide analysis. This study showed that ChromoGen’s produced structures are transferable and validated their generic physical properties.
Transferability of chromogens between cell types
A DNA accessibility model called ChromoGen used DNase-seq data to predict conformational ensembles with cell type specificity. During cell development, chromatin changes in three dimensions: DNA accessibility and epigenetic alterations. IMR-90 cells were used to evaluate the model using the hg19 sequence and their DNase-seq data. With a median value of 0.9533, the results demonstrated a strong correlation between ChromoGen distances and the logarithm of contact probabilities calculated from population Hi-C data. The application of ChromoGen to other cell types is supported by the quality of the anticipated IMR-90 values, which are equivalent to those found for GM12878 cells.
Conclusion
ChromoGen is a computational method that makes de novo predictions about chromatin conformations using DNA sequence data. Its generative nature eliminates the need for polymer simulations’ expensive state space navigation and enables the rapid generation of statistically independent samples. To extract characteristics from sequencing data and place them in low-dimensional embeddings that the diffusion model can handle effectively, ChromoGen uses a transformer-based front end. By lowering the computational cost of each diffusion step, this design offers a workable way to obtain de novo predictions specific to a cell type while fully utilizing DNA sequence and chromatin accessibility information. Any cell type with available DNAse-seq data can use ChromoGen, allowing for various investigations into the variations in genome organization within and between cell types.
Article Source: Reference Paper | Reference Article | The source code for ChromoGen is available on Zenodo and GitHub.
Disclaimer:
The research discussed in this article was conducted and published by the authors of the referenced paper. CBIRT has no involvement in the research itself. This article is intended solely to raise awareness about recent developments and does not claim authorship or endorsement of the research.
Follow Us!
Learn More:




Deotima is a consulting scientific content writing intern at CBIRT. Currently she's pursuing Master's in Bioinformatics at Maulana Abul Kalam Azad University of Technology. As an emerging scientific writer, she is eager to apply her expertise in making intricate scientific concepts comprehensible to individuals from diverse backgrounds. Deotima harbors a particular passion for Structural Bioinformatics and Molecular Dynamics.











{kind=link}