
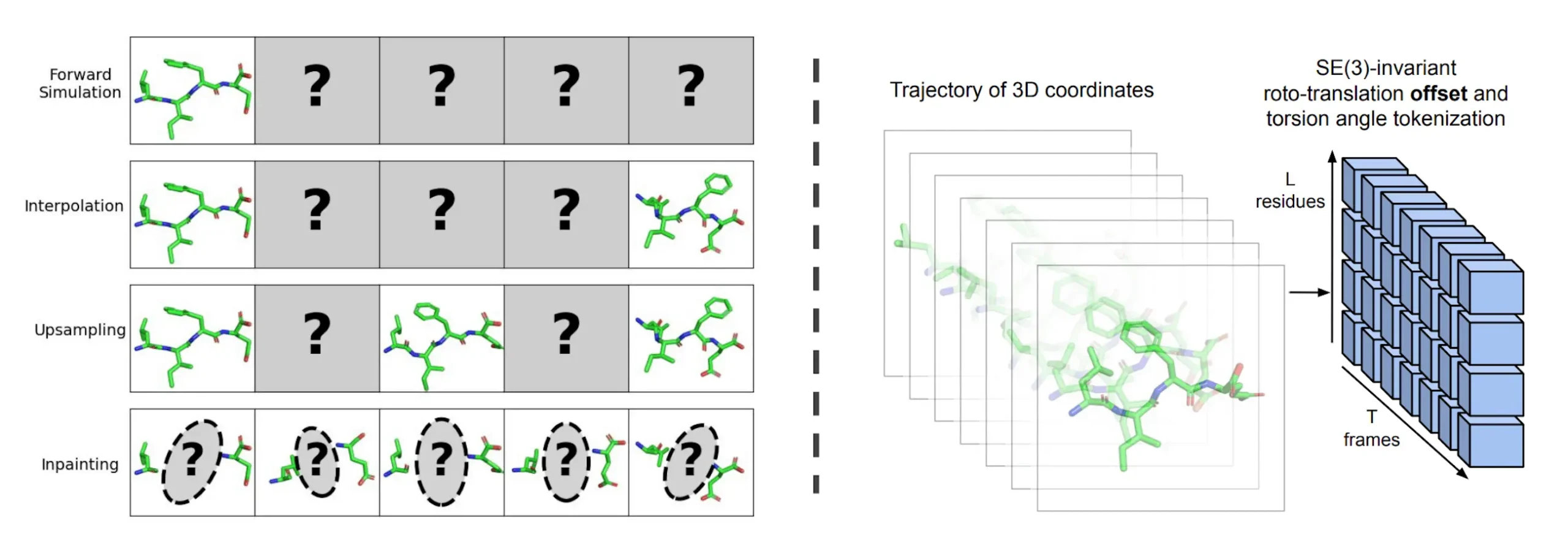
As a paradigm for developing adaptable multi-task surrogate models of MD from data, researchers from the Massachusetts Institute of Technology present generative modeling of molecular trajectories. Researchers demonstrate how such generative models can be tailored to a variety of tasks, including forward simulation, transition path sampling, and trajectory upsampling, by conditioning on carefully selected frames of the trajectory. Researchers also illustrate the initial stages of dynamics-conditioned molecular design by conditioning a portion of the molecular system and inpainting the remainder. Researchers demonstrate that the model is capable of producing appropriate ensembles of protein monomers by validating the entire range of these capabilities using tetrapeptide simulations.
Introduction
A popular method for examining various molecular events in chemistry, biology, and other molecular sciences is molecular dynamics (MD), which is the numerical integration of Newton’s equations of motion at atomic sizes. Despite being generic and flexible, MD requires a lot of computing power since integration processes and pertinent chemical phenomena occur on quite different timelines. Thus, the goal of a large body of research spanning several decades is to improve or speed up MD simulation algorithms’ sampling efficiency. In the last few years, deep generative modeling research has focused on learning surrogate models of MD. Nevertheless, current training paradigms only apply to a small number of downstream problems because they cannot properly utilize the abundant dynamical information in MD training data.
Insights into MDGEN: A Generative Modeling Paradigm for Molecular Trajectories
In this paper, researchers provide MDGEN, a novel paradigm based on direct generative modeling of simulated trajectories, enabling quick, general-purpose surrogate modeling of MD. Researchers formulate end-to-end generative modeling of entire trajectories viewed as time series of 3D molecule structures, in contrast to earlier research that learned the autoregressive transition density or equilibrium distribution of MD. The formulation of the problem adds a time dimension to single-structure generative models, similar to how image generative models were extended to videos. This allows the model to be used for a wider range of forward and inverse problems. These generative models function as well-known surrogate forward simulators of the reference dynamics when given (and dependent upon) the initial “frame” of a particular system.
Inverse problems in molecular systems can be effectively addressed by generative models, particularly molecular video generative models. Upsampling, inpainting, interpolation, and forward simulation are all made possible by these models. To sample a realistic path between two endpoints, interpolation is used, whereas forward simulation samples a possible time evolution of the molecular system. To obtain a trajectory with timestep ∆t/M, upsampling upsamples the “framerate” and uses trajectories saved at less frequent intervals to infer fast motions. By generating the remaining portion of a molecule and its time evolution to match the known portion of the trajectory, inpainting enables the creation of molecules to scaffold desired dynamics.
Limitations
- The model’s primary drawback is its dependence on keyframes, which prevents it from unconditionally generating or inpainting residual roto-translations.
- The architecture might not be the most appropriate for bigger motions, as evidenced by the poorer performance of proteins compared to peptides.
- Similar to the content-frame decomposition of video diffusion models, the co-generation of keyframes and trajectory tokens can be achieved by fine-tuning single-structure models.
- Alternative tokenization techniques will be required for simulating the trajectories of more diverse and generic systems, such as chemical ligands, materials, or explicit solvents, in addition to proteins and peptides.
- The capacity to represent trajectories over a region of space where atoms may enter and exit, rather than a predetermined set of atoms, may be necessary for more ambitious applications.
Opportunities
MD trajectory generation may be a multifunctional, unifying paradigm for deep learning over the microscopic environment, much as the fundamental function of video generative models for comprehending the macroscopic world. In a broader sense, interpolation is the process of generating hypotheses regarding the mechanisms underlying arbitrary molecular occurrences, particularly when only a portion of the end states are known. Redesigning proteins to enhance rare transitions seen only once in a simulation or (with ab initio trajectories) de novo design of enzymatic mechanisms and motifs are two examples of how molecular inpainting may be used as a general technique to design molecular machinery by scaffolding more intricate and fine-grained dynamics.
Conclusion
Deep learning-based surrogate models are becoming more popular due to the computational cost of molecular dynamics (MD), even though MD is a very effective method for understanding microscopic phenomena. Molecular trajectory generative modeling is presented as a framework for learning adaptable multi-task surrogate models of MD from data. It is possible to adapt generative models to a variety of tasks, including forward simulation, transition path sampling, and trajectory upsampling, by conditioning on frames of the trajectory. Altogether, the study shows how generative modeling may extract value from MD data for various downstream tasks that are difficult to handle with current techniques or even MD itself. Additional studies may become possible in the future as ground truth MD trajectory data for many chemical systems become available. Furthermore, there may be opportunities for theoretical investigation due to factors specific to molecular trajectories, such as Markovianity, equilibrium versus non-equilibrium processes, and the reversibility of the microscopic world in contrast to the physical world.
Article Source: Reference Paper | Reference Article | Code is available at GitHub.
Disclaimer:
The research discussed in this article was conducted and published by the authors of the referenced paper. CBIRT has no involvement in the research itself. This article is intended solely to raise awareness about recent developments and does not claim authorship or endorsement of the research.
Important Note: arXxiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Follow Us!
Learn More:




Deotima is a consulting scientific content writing intern at CBIRT. Currently she's pursuing Master's in Bioinformatics at Maulana Abul Kalam Azad University of Technology. As an emerging scientific writer, she is eager to apply her expertise in making intricate scientific concepts comprehensible to individuals from diverse backgrounds. Deotima harbors a particular passion for Structural Bioinformatics and Molecular Dynamics.











{kind=link}