
In a recent breakthrough in AI-driven drug discovery, researchers from Pfizer, AstraZeneca, and Chalmers University unveiled a new generative framework called FLOWR, designed to revolutionize structure-based ligand generation. Their collaborative work bridges machine learning, computational chemistry, and pharmaceutical research and is set to address some of the most persistent challenges in early-stage drug design.
Why Structure-Based Drug Design Needs a Boost
From the outset of rational drug design, Structure-based drug discovery (SBDD) has served as a pillar, utilizing a drug’s 3D protein structure to inform its development. SBDD has traditionally relied on computational molecular docking and virtual screening methods. While productive, these methods often fail to yield accurate predictions, resulting in highly inefficient and costly attempts at drug development due to the endless spaces that contain complications arising in the interactions between the drug molecules and the target proteins.
With the latest developments in deep learning, model-based methods such as diffusion models are now beginning to affect this field. Diffusion models have the ability to generate new molecules by following a certain algorithm, wherein the required structures are synthesized through noise that is shaped into chemical formations, and protein binding sites are used as a framework. However, because these models generate slow results, integrate non-viable structures, and can be easily modified, their usefulness is drastically lowered.
FLOWR: A New Generation of Structure-Aware Ligand Design
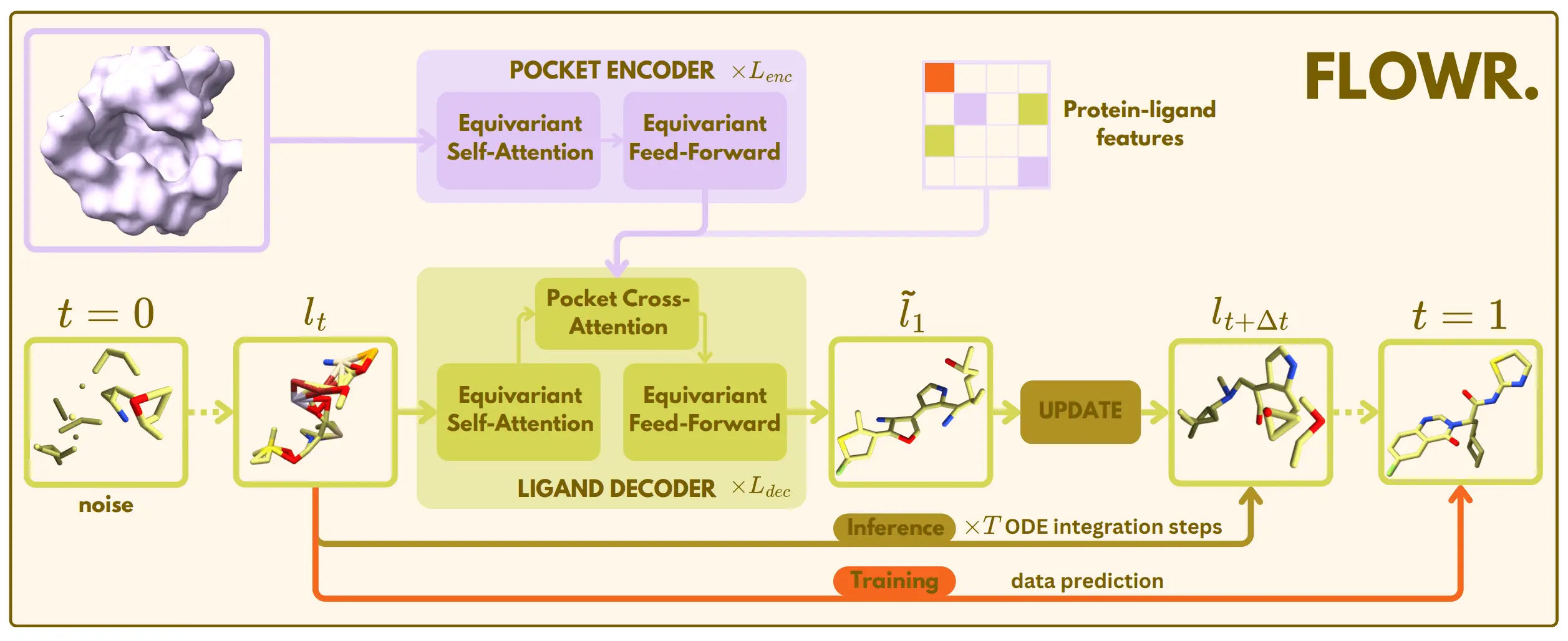
FLOWR (Flow Matching for Structure-Aware De Novo, Interaction- and Fragment-Based Ligand Generation) is a novel generative model that takes a different approach. Instead of relying on slow, iterative diffusion processes, FLOWR uses flow matching—a technique that learns efficient, direct transformations from noise to data. This allows it to generate three-dimensional ligand structures much faster and with greater accuracy.
What makes FLOWR unique is that it can control ligand invention based on the spatial intricacies of protein pockets in 3D, something no other system has achieved so far. FLOWR possesses a specialized protein pocket encoder that quantitatively captures the geometry and chemistry of the binding site. Thus, it guarantees that generated molecules are not merely validated from a chemical perspective but will actually have the right shape and smaller chemicals to bind favorably to the protein.
Introducing SPINDR: A Gold-Standard Dataset
The quality of existing datasets has always been a significant hurdle in training and evaluating generative models for SBDD. Popular datasets CrossDocked2020 and PDBBind, for example, include missing atoms, unrealistic poses of the ligands, and even data leakage between training and test datasets. These disabilities often yield models that work perfectly during performance benchmarks but fail miserably when put through practical drug discovery workflows.
In an attempt to remediate this, SPINDR (Small Molecule Protein Interaction Dataset, Refined) was created by the team; it is a dataset that has been meticulously constructed from crystallographic data. SPINDR is equipped with over 35,000 protein-ligand complexes that are high in quality, fully trimmed to energetic equilibrium, have explicit hydrogens, and possess intricate interaction fingerprints. The extensive filtering applied guarantees the lack of excessive redundancy and information leakage, making SPINDR the new benchmark for models designed to provide optimal training and evaluation results.
Real-World Impact: From Hit Expansion to Lead Optimization
The diagnostic capabilities of FLOWR and FLOWR.multi have been showcased in relevant targets, including lipoprotein-associated phospholipase A2 (5YEA) and pyruvate dehydrogenase kinase (4MPE). In these case studies, the models yielded comprehensively balanced sets of ligands that not only matched the known bioactivity and chemical constituents of the inhibitors but also provided additional opportunities for chemical modifications.
Furthermore, the constructed molecules performed exceptionally on the drug-likeness and synthetic accessibility parameters, which proves that the generated results with FLOWR are not simply of theoretical value but indeed beneficial in actual drug development processes.
Looking Ahead: A New Standard for AI-Driven Drug Design
Both FLOWR and SPINDR mark a colossal leap in applying AI for drug design at the molecular level. Incorporating advanced generative modeling coupled with the meticulous curation of datasets allows the authors to create robust but useful tools. With FLOWR’s speed and accuracy and FLOWR.multi’s flexibility integrated into ligand synthesis, FLOWR shall set off a new period of prompt, exact, and dependable ligand construction.
As the field moves forward, the adoption of high-quality datasets like SPINDR and versatile models like FLOWR will be crucial for translating AI advances into real therapeutic breakthroughs. The future of drug discovery is not just faster and smarter—it’s FLOWR-powered.
Article Source: Reference Paper
Disclaimer:
The research discussed in this article was conducted and published by the authors of the referenced paper. CBIRT has no involvement in the research itself. This article is intended solely to raise awareness about recent developments and does not claim authorship or endorsement of the research.
Important Note: arXiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Follow Us!
Learn More:




Anchal is a consulting scientific writing intern at CBIRT with a passion for bioinformatics and its miracles. She is pursuing an MTech in Bioinformatics from Delhi Technological University, Delhi. Through engaging prose, she invites readers to explore the captivating world of bioinformatics, showcasing its groundbreaking contributions to understanding the mysteries of life. Besides science, she enjoys reading and painting.











{kind=link}