
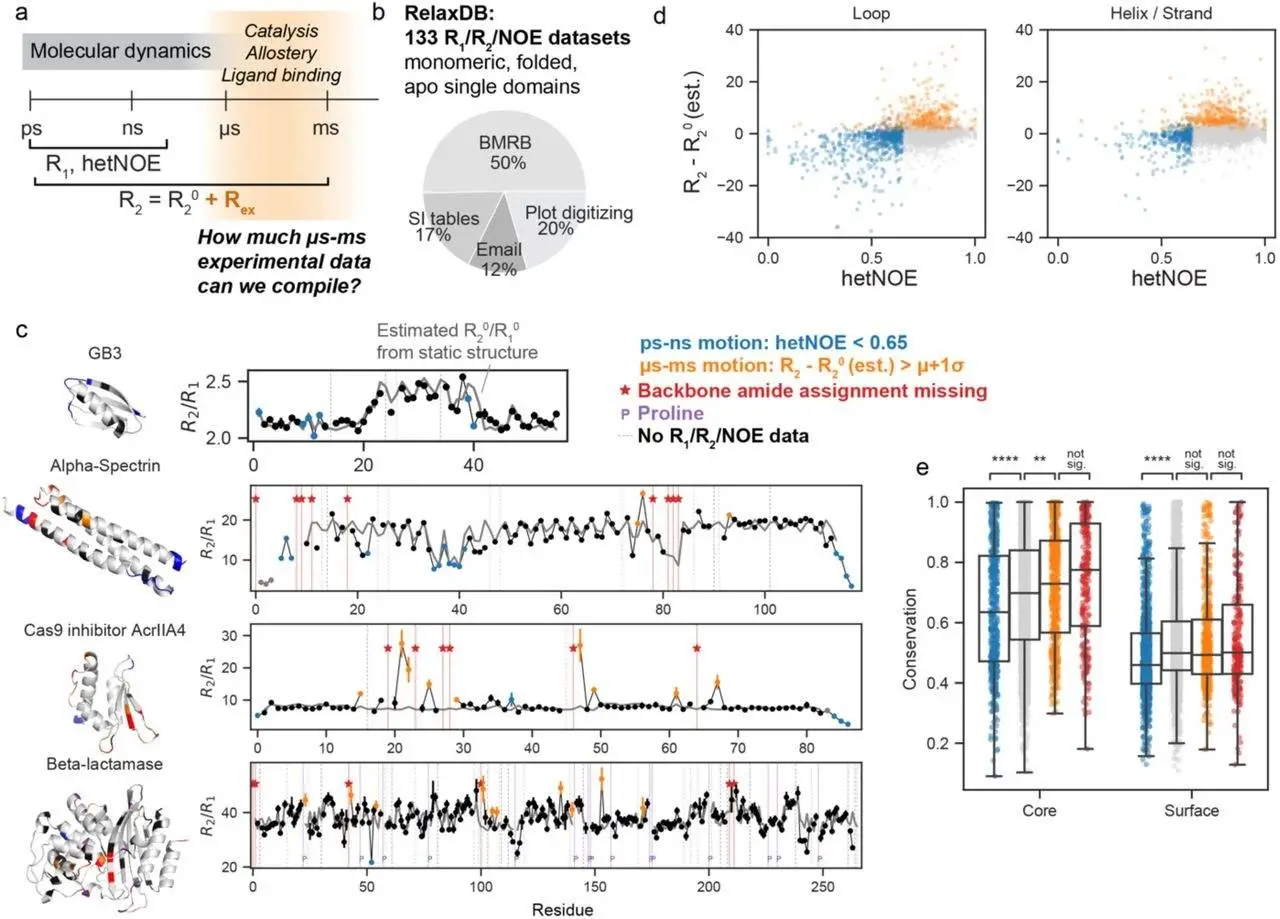
There are more than 100 NMR relaxation datasets in the Biological Magnetic Resonance Data Bank (BMRB), which show an observable motion in µs-ms. NMR signals can be broadened beyond detection by millisecond motions, leaving residues unassigned in chemical shift datasets of about 10,000 proteins. Deep learning models were taught to predict missing assignments caused by µs-ms motions. These models also predict µs-ms motion that is directly detected in NMR relaxation experiments. Future research depends on this accomplishment in understanding protein interactions and functions at micro- to millisecond timescales. The multimodal language model ESM-32’s intermediate layer is utilized by the best of these models, which researchers from Scripps Research and Howard Hughes Medical Institute have called Dyna-1. It is particularly good at predicting dynamics that are directly related to biological function, such as ligand binding and enzyme catalysis, which is consistent with our findings that residues with µs-ms motions are highly conserved. Researchers believe the datasets and models described here will be revolutionary in revealing the common language of dynamics and function.
Introduction
The activities of proteins rely on their capacity to interconvert between different conformations, and a better knowledge of basic biological systems would result from an increased capacity to anticipate various conformations and timelines. Although there has long been a desire to describe the motions of proteins since molecular dynamics simulations computationally, the prediction power of single structures is not as high as that of proteins. When combined with extensive sequencing data and contemporary deep-learning architectures, AlphaFold-2 (AF2) showed that experimental data from the Protein Data Bank might produce previously unheard-of levels of accuracy in practical tasks. The lack of large-scale, defined benchmarks of experimental observables in protein dynamics prediction, however, presents a challenge for computational approaches, whether deep learning-based or simulation-based, as there is a dearth of standardized experimental data on dynamics to assess and train on.
Paper’s Work Using NMR Observables
The work illustrated the predictive ability of utilizing a basic NMR observable for dynamics on a scale of roughly 10,000 proteins in the event that a residue 15N backbone assignment is absent. Compared to other indicators of heterogeneity in X-ray and EM structures, this observable is unique. Despite problems with the existing publically available data, the researchers were able to get labels for their spectra, which are the final output of processing NMR data. For science, the absence of uniform standards for depositing the spectra is a major setback. A set of 100 raw spectra, two orders of magnitude smaller than the current dataset, was selected by the researchers.
Future developments in machine learning techniques may include automated assignment algorithms, higher throughput and diversity samples, and purposefully lower resolution data to gather information on exchange-broadened peaks and peak widths swiftly. The BMRB’s metadata labeling could use some work. Understanding the dynamics in both situations would surely be enhanced by combining in vitro structural data with high-throughput readouts of fitness data.
About Deep Learning Model Dyna-1
Dyna-1, the developed model, can predict physiologically relevant µs-ms motions based on measurements of µs-ms motion collected by numerous separate labs on proteins that have been described using various types of studies. Notably, our approach caused the published NMR relaxation data to be reinterpreted in a number of ways. Researchers discovered that some proteins interact with phosphate ions as part of their biological function while looking into “false negatives” in the RelaxDB dataset. However, the NMR relaxation studies were carried out in a phosphate buffer. Phosphate binding did not predict exchange in Dyna-1. On the other hand, Dyna-1 did anticipate millisecond motions that were overlooked by conventional analysis of Carr-Purcell-Meiboom-Gill (CPMG) studies; nevertheless, this was confirmed by a closer examination of the available NMR data.
Architecture of Dyna-1
Dyna-1’s foundational ESM-3 was trained using C-alpha structure data rather than sidechain data. Performance may be enhanced by including sidechain-aware training. Furthermore, Dyna1 is only trained on proteins with fewer than 400 residues, which may restrict its ability to forecast the dynamics of larger proteins. Research is still being conducted to determine how sensitive Dyna-1 is to dynamics differences within the same family and how well it can predict the dynamics of de novo proteins. There was only a slight improvement in performance when researchers compared the Dyna-1 model architecture with a training split of 30% sequence identity, 0.5 TM-score cutoff, to a more relaxed training split of 80% sequence identity, 1.0 TM-score cutoff.
Achievements of Dyna-1
The α2-β4 loop has high p(missing), as predicted by Dyna-1, but the majority of these residues are allocated. Our only source of ground truth data on µs-ms motion would be the missing peak label, which would result in a large number of false positives. It was challenging to assign the α2-β4 loop and analyze the relaxation data that followed; the authors explained, “because of severe line broadening,” which is blatant evidence of µs-ms exchange. So that researchers could better understand the predictive power of the model, they had to test Dyna-1’s predictions using the more informative relaxation data in RelaxDB.
The biological significance of dynamic motifs in bigger proteins is revealed by Dyna-1’s correct prediction of these motifs. It forecasts significant exchange in the FMN ligand-binding region of flavodoxin YcqA. The human olfactory marker protein predicts a high p(exchange) in the omega-loop, which is highly conserved and has a significant Rex. A nuclear export signal is produced by the omega loop. Additionally, Dyna-1 forecasts high p(exchange) in APOBEC-2, which regulates the selectivity of DNA substrates.
The protein MAP kinase p38 gamma (MK12) is extremely dynamic, with ps-ns and µs-ms processes taking place all over its structure. Future advancements in the ability to distinguish between ps-ns and µs-ms motions are indicated by Dyna-1 projections. Dyna-1’s highest p(exchange) is found in regions with ps-ns mobility in several proteins. It is difficult to interpret these predictions, though, because residues undergo both motions, and one field strength is not enough to measure motions at two timescales at once.
Conclusion
Protein structure prediction was made possible by AlphaFold-2 (AF2), which transformed molecular biology. This was made possible by the Protein Data Bank (PDB) and the Critical Assessment for Structure Prediction (CASP) challenges. Though experimental measurements have resisted being gathered into standardized databases, a comprehension of protein dynamics is crucial to an understanding of protein function. The exchange and deposit of experimental data on macromolecule dynamics is essential because of Dyna-1’s predictive capability. A predictive model can be trained on various experimental data sources, such as missing assignments, Rex from R1/R2/NOE, and numerous exchange indications from CPMG, as this work shows.
Article Source: Reference Paper | Dyna-1 code is available for noncommercial use on GitHub.
Disclaimer:
The research discussed in this article was conducted and published by the authors of the referenced paper. CBIRT has no involvement in the research itself. This article is intended solely to raise awareness about recent developments and does not claim authorship or endorsement of the research.
Important Note: bioRxiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Follow Us!
Learn More:




Deotima is a consulting scientific content writing intern at CBIRT. Currently she's pursuing Master's in Bioinformatics at Maulana Abul Kalam Azad University of Technology. As an emerging scientific writer, she is eager to apply her expertise in making intricate scientific concepts comprehensible to individuals from diverse backgrounds. Deotima harbors a particular passion for Structural Bioinformatics and Molecular Dynamics.











{kind=link}