
A major factor in evolution, genome duplications have different effects on the expression, function, and regulation of genes. However, multiple duplication modes make it difficult to detect and categorize duplicated genes because of antiquated or useless instruments. Here, scientists from Ghent University, University of Pretoria, and Nanjing Agricultural University introduce doubletrouble, an R/Bioconductor program that offers a thorough and reliable framework for examining genomic data for duplicate genes. The six duplication modes—segmental, tandem, proximal, retrotransposon-derived, DNA transposon-derived, and dispersed duplications—can be used by doubletrouble to identify and categorize gene pairs. It can also compute substitution rates, identify signatures of suspected whole-genome duplication events, and display the results as illustrations suitable for publication. In 822 eukaryotic genomes, researchers used doubletrouble to categorize the duplicated gene repertoire. The results were made accessible via an intuitive web interface.
Introduction
Duplications of genes and genomes provide evolution with a significant amount of new genetic material. Yet, the ways in which gene and genome duplications contribute to genome evolution differ, and genes produced by these two processes exhibit distinct evolutionary trajectories at the transcriptome, epigenomic, and genomic levels, as well as evolving under various selection pressures. Additionally, based on the mechanism of duplication, gene retention may be skewed, with some gene classes—such as transcription factors and protein complex members—being preferentially preserved during whole-genome duplication.
Understanding doubletrouble
Researchers present double trouble, an R/Bioconductor program for locating, categorizing, and examining genes that are duplicated in genomic data. doubletrouble was created to readily interface with other Bioconductor packages because it is a component of the Bioconductor ecosystem of R packages.
Gene pairs that are duplicated can be recognized and categorized by doubletrouble as originating from segmental, tandem, proximal, retrotransposon-derived, DNA transposon-derived, and scattered duplications. Calculating substitution rates (Ka, Ks, and their ratio Ka/Ks), looking into the signatures of possible whole-genome duplication events, and producing figures suitable for publishing can all be done with duplicate pairs found and categorized with double difficulty. By recognizing and classifying the complete repertoire of duplicated genes in 822 eukaryotic genomes from Ensembl examples, researchers show the efficacy of doubletrouble. Lastly, they developed a web application (https://almeidasilvaf.github.io/doubletroubledb) that allows users to examine and download data gathered here on the repertoire of duplicated genes across the Eukarya tree of life to ease data reuse.
Execution of doubletrouble
Gene annotations as GRanges objects and whole-genome protein sequences (one per gene, primary transcript only) as AAStringSet objects are the necessary input data. Both must be in lists with matching names that indicate species names or other genome identifiers. With the help of the R package syntenet’s fasta2AAStringSetlist() and gff2GRangesList() functions, users can generate such lists of AAStringSet and GRanges objects from several FASTA and GFF/GTF files in a directory. The syntenet package’s process_input() function must be used to process the sequence and annotation data to ensure that each gene has only one sequence.
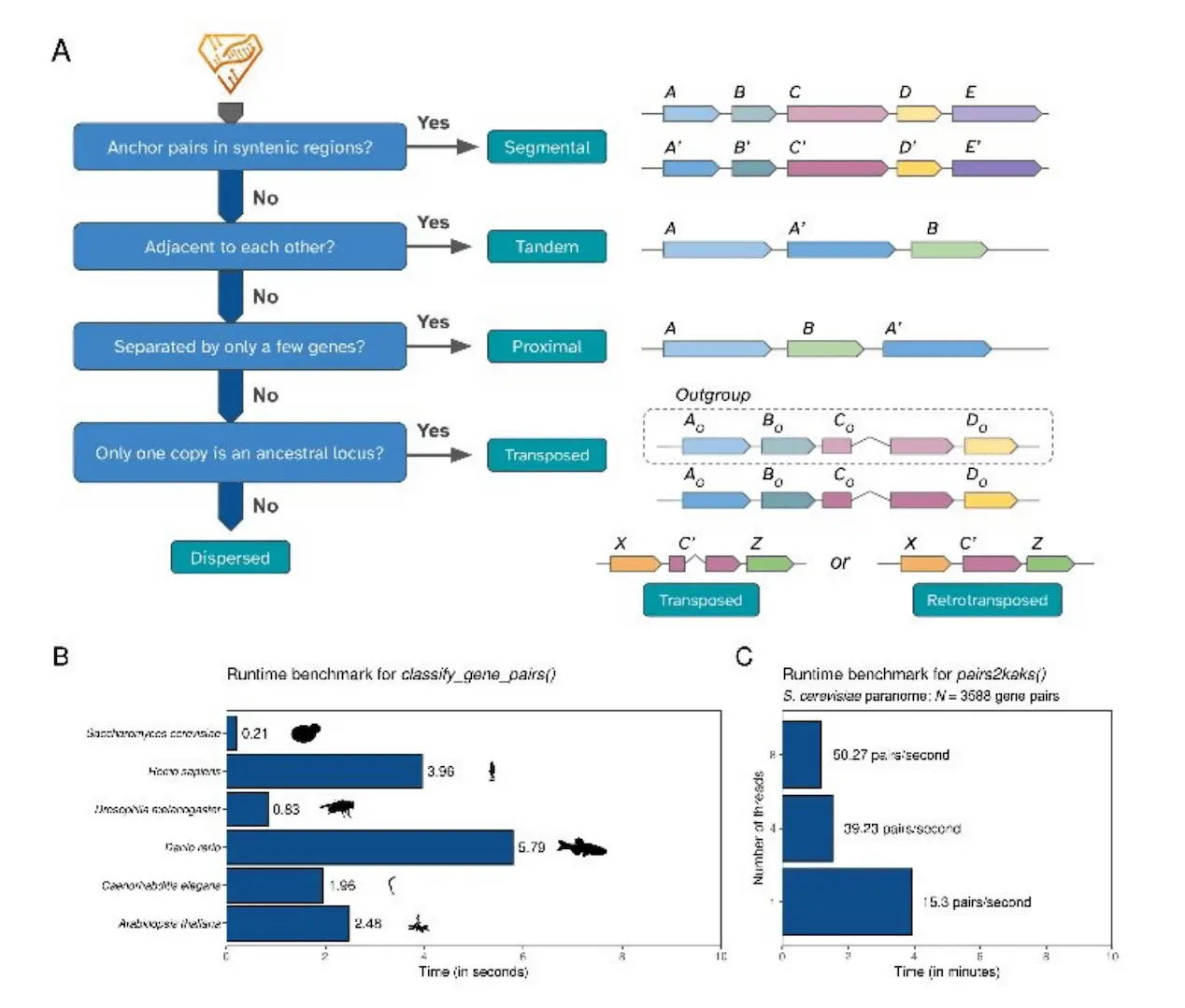
A function called run_diamond() is provided by the syntenet package to do intraspecies similarity searches using DIAMOND. By default, top hits = 5 and Evalue = 1e-10 are used. The read_diamond() method allows users to read the tabular output of processed sequences and export them to FASTA files. While all paralogous gene pairings in a genome are included in the tabular output, users can further limit the findings by coverage, similarity, etc. The method classify_gene_pairs() takes genes and their annotations as input and uses four schemes—binary, standard, extended, and complete—to categorize paralogs according to their form of duplication.
Paralogs are categorized by duplication mode using the syntenet software, which also finds intragenomic syntenic areas. Segmental duplicates (SD), small-scale duplicates (SSD), tandem duplicates (TD), proximal duplicates (PD), and distributed duplicates (DD) are the several types of paralogs found in syntenic regions. Users must supply a similarity search table between all query species and an outgroup to categorize DD pairs as coming from transposon-derived duplications (TRD). DD pairings with more than one species are classified as TRD, whereas those with only one copy having an ancestral locus are categorized as transposed duplicates (TRD). DNA transposon-derived duplications (dTRD) and retrotransposed duplicates (rTRD) are also classified. The classify_genes() program uses the hierarchy SD > TD > PD > rTRD > dTRD > DD to assign genes to distinct duplication modes.
Using 17 codon-based models from KaKs_Calculator 2.0, the method “pairs2kaks()” determines the rates of synonymous substitution per substitution site (Ks), nonsynonymous substitutions per substitution site (Ka), and their ratios for every gene pair. It is possible to differentiate between duplications of broad genomic areas and segmental duplicates that most likely occurred from WGD events using parameters of mixture components and age groups.
Limitation
Using parameters acquired with DIAMOND (or other sequence similarity search algorithms), doubletrouble categorizes duplicate pairs according to duplication mode. Nevertheless, in organisms with frequent horizontal gene transfers (such as prokaryotes), finding paralogs based on sequence similarity may result in false positives. Therefore, in these situations, it is advised to use techniques that can differentiate between paralogs and xenologs (homologs resulting from horizontal gene transfer), followed by the use of paralogs-only for classification with twofold problems.
Conclusion
doubletrouble is a tool for R/Bioconductor that may be used to compute substitution rates for gene pairs, locate and categorize duplicate genes by duplication mechanism, and produce figures that are appropriate for publishing. This package should aid in investigating the duplication landscape across many species, and the data sets produced by this investigation will be a valuable tool for scientists looking into how gene duplication evolved in eukaryotes.
Article Source: Reference Paper | doubletrouble is available on GitHub | Website.
Disclaimer:
The research discussed in this article was conducted and published by the authors of the referenced paper. CBIRT has no involvement in the research itself. This article is intended solely to raise awareness about recent developments and does not claim authorship or endorsement of the research.
Follow Us!
Learn More:




Deotima is a consulting scientific content writing intern at CBIRT. Currently she's pursuing Master's in Bioinformatics at Maulana Abul Kalam Azad University of Technology. As an emerging scientific writer, she is eager to apply her expertise in making intricate scientific concepts comprehensible to individuals from diverse backgrounds. Deotima harbors a particular passion for Structural Bioinformatics and Molecular Dynamics.











{kind=link}