
G protein-coupled receptors (GPCRs) are essential membrane proteins with dynamic conformations that are also major targets for drug development and discovery. However, designing protein agonists and antagonists has proven difficult. In this article, researchers outline computational de novo design techniques and a high throughput “receptor diversion” microscopy-based screen for producing GPCR binding miniproteins with high affinity, potency, and selectivity. These techniques are then used to produce MRGPRX1 agonists and CXCR4, GLP1R, GIPR, GCGR, and CGRPR antagonists. The precise conformational control of receptor activity is confirmed by cryo-electron microscopy data, which shows atomic-level agreement between the planned and experimentally determined structures for MRGPRX1-bound agonists and CGRPR-bound antagonists. New avenues for GPCR drug development and discovery are made possible by our de novo design and screening methodology.
Introduction
The human genome has the largest and most varied family of membrane receptors, known as G protein-coupled receptors (GPCRs), which are essential for numerous physiological functions. GPCRs are at the forefront of drug discovery and development because they are linked to various illnesses, such as cancer, metabolic and cardiovascular conditions, and neurological disorders. Over the past decades, biologics like antibodies, nanobodies, and peptides have gained momentum as GPCR treatments and tools. The creation of biologics that modulate GPCR signaling is still a difficult task, nevertheless, and frequently calls for a combination of approaches like screening random libraries or inserting peptide snippets from natural proteins. Generating GPCR agonists has proven extremely challenging, requiring significant efforts in antibody and receptor engineering.
Many biologically significant targets can now have miniprotein binders designed with atomic-level precision thanks to developments in computational protein design. The design of miniprotein binders with desired characteristics, such as remarkable selectivity, high protease stability, and prolonged biological half-life, is made possible by methodologies like RFdiffusion and Rosetta. Despite these developments, there are still many obstacles to overcome, especially for functional miniproteins that target membrane-embedded binding pockets like flexible, recessed GPCR epitopes, which require conformational specificity to induce function. Researchers decided that in order to address these issues, new high-throughput experimental screening techniques and specialized computational design would be required, so researchers set out to create suitable techniques.
Researchers Developed Computational Methods to Target GPCR Epitopes
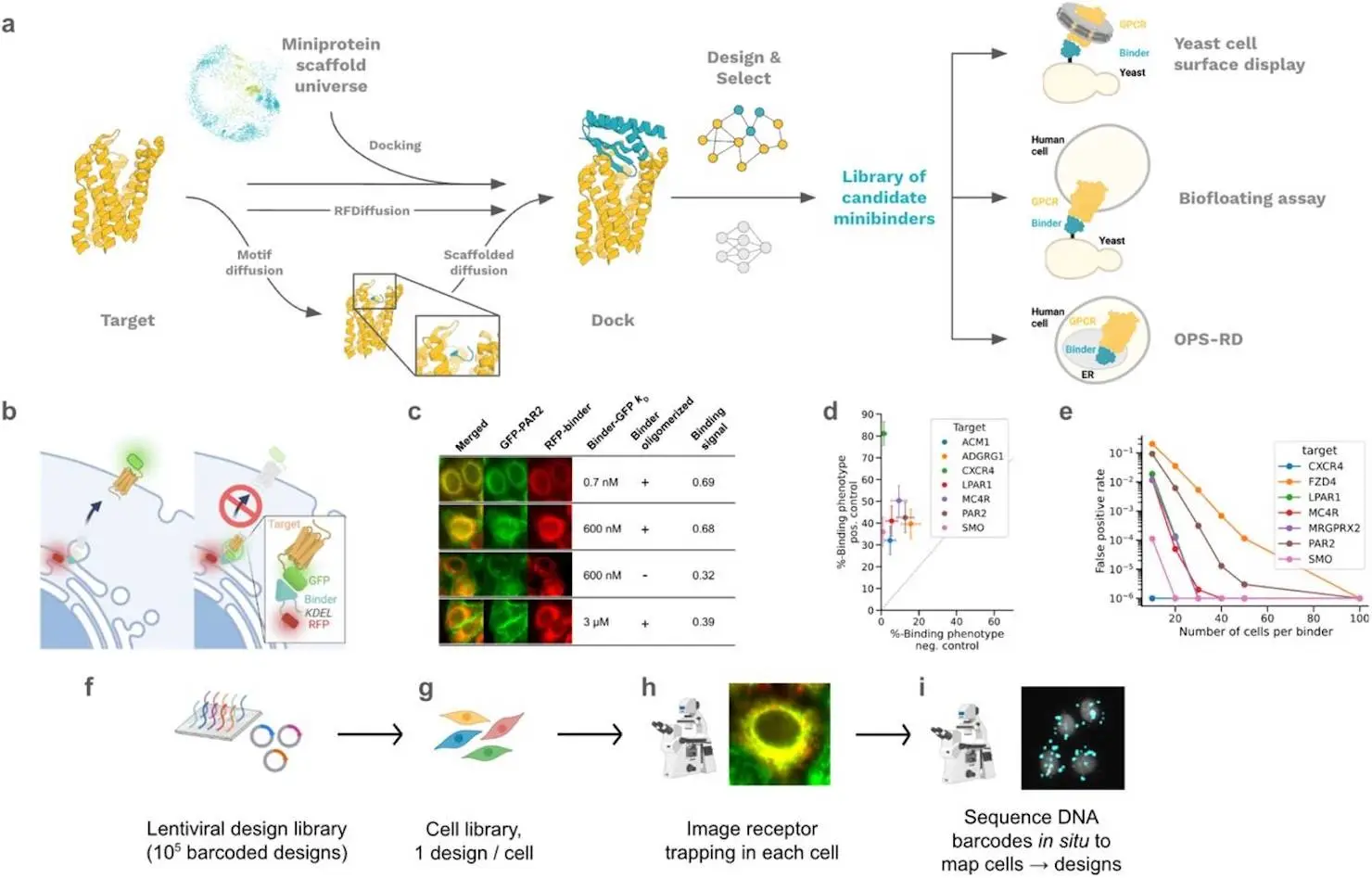
To increase the diversity of designs for library-scale experimental screening, researchers first developed a “motif-directed” RFdiffusion approach that, instead of diffusing an entire binding protein, starts with just a five-residue peptide (the motif) to interact with target hot spot residues within the recessed binding pocket. This allows the short peptide to more easily penetrate into the deep pocket. Once good solutions for a binding peptide are found, the interacting peptide is kept in a fixed position, and full mini proteins are generated using the motif-scaffolding capabilities of RFdiffusion.
Second, researchers created a method called MetaGen that uses structurally varied scaffolds from the metaproteome produced by AlphaFold in Rosetta RifDock computations. Unlike conventional de novo miniprotein backbone libraries, which are frequently made up of short loops and straight helices that are not suitable for binding deeply recessed epitopes, these scaffolds have protruding elements like beta-hairpin loops and kinked helices. However, they can still be reliably predicted from a single sequence, which is a crucial designability criterion. Researchers employed ProteinMPNN for sequence design, AlphaFold2 (AF2) initial guess, and Rosetta metrics for filtering designs after backbone design using either RFdiffusion or MetaGen. In a similar manner, researchers used the MetaGen backbone library or created backbones from scratch using RFdiffusion to design class B receptor antagonists.
Given the difficulty of class A GPCR epitopes, researchers reasoned that robust discovery of functional binders would require the use of high-throughput screening (HTS) techniques in addition to computational design. To solve this, researchers created Receptor Diversion (RD), an HTS assay that works directly in human cells without the need for purification. The potential binder and the membrane protein target are both expressed in a human cell line for this experiment, and the binder is located inside the secretory route using a genetic tag (such as an endoplasmic reticulum (ER) retention signal). This enables interaction between the binder and the membrane protein target’s extracellular face. The target is “diverted” from its typical trafficking pattern by high-affinity contacts, which is represented by an increase in binder-target colocalization.
Researchers found a strong binding signal appropriate for high-throughput screening across seven different GPCRs, with a cross-GPCR Z′ average of 0.47 when sampling 100 cells per binder. The advantages of RD are as follows: (i) the target can be expressed at near-endogenous levels in a relevant cell line and does not need to be produced as a stable soluble protein, which is difficult for GPCRs, as required for display methods; (ii) binders found through the screen must be soluble and function in the molecularly crowded environment of the secretory pathway, and (iii) the binder must specifically bind the target in order to induce receptor diversion. With the RD platform, up to 100,000 designs can be screened by imaging up to 107 cells and obtaining single-cell expression and co-localization data. At the outset of this investigation, researchers were uncertain about the practicality of RD screening for library screening, therefore researchers also investigated the use of yeast display in conjunction with either soluble GPCRs in nanodiscs or GPCRs shown on mammalian cells (biofloating).
Conclusion
Drug discovery is made extremely difficult by the dynamic nature of GPCRs because of their structural complexity. These issues can be resolved by de novo design, which produces miniprotein binders with a variety of affinity, potency, and selectivity profiles that target MRGPRX1, CXCR4, GLP1R, GIPR, GCGR, and CGRPR. Agonists have been especially difficult to find because they must be conformally selective, meaning they must distinguish between minute structural variations in the orthosteric binding site. There are now two atomically precise MRGPRX1 binders—full and partial miniprotein agonists—that can accurately manipulate the receptor’s structure to cause agonism. This demonstrates that de novo design is a practical method for creating GPCR-targeting ligands that can identify and precisely regulate a conformal epitope to produce a specific and intended pharmacological effect. GPCR agonists and antagonists that are de novo developed have significant therapeutic potential because of their crucial roles in both illness and cellular function. This study opens the door for revolutionary GPCR-related applications in drug development and fundamental research.
Article Source: Reference Paper
Disclaimer:
The research discussed in this article was conducted and published by the authors of the referenced paper. CBIRT has no involvement in the research itself. This article is intended solely to raise awareness about recent developments and does not claim authorship or endorsement of the research.
Important Note: bioRxiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Follow Us!
Learn More:




Deotima is a consulting scientific content writing intern at CBIRT. Currently she's pursuing Master's in Bioinformatics at Maulana Abul Kalam Azad University of Technology. As an emerging scientific writer, she is eager to apply her expertise in making intricate scientific concepts comprehensible to individuals from diverse backgrounds. Deotima harbors a particular passion for Structural Bioinformatics and Molecular Dynamics.











{kind=link}