
The GPCR Dock competitions are a set of community-wide evaluations of ligand docking and computational structural modeling for G protein-coupled receptors, a key class of therapeutic targets in the human proteome. To shape and guide the development of computational modeling approaches for GPCRs, the assessments are intended to offer an objective summary of advancements and identify areas that require improvement. The fourth round (GPCR Dock 2021), which followed in the footsteps of the 2008, 2010, and 2013 evaluations, included five unique and difficult prediction targets and came at the same time as AlphaFold, the ground-breaking Artificial Intelligence (AI) technology for predicting protein structures from amino acid sequences. Researchers from the University of California and ShanghaiTech University show how advances in AI-powered modeling have made it possible for contemporary computational models of GPCR complexes with peptides to not only match but even surpass low-resolution actual structures in terms of accuracy. The findings, however, demonstrate the absolute necessity of high-resolution GPCR structure identification, particularly when working with small-molecule compounds, and the simultaneous use of expert-guided and physics-based modeling techniques.
Introduction to GPCRs and the GPCR Dock Competitions
A key class of cell membrane receptors in the human proteome, the G protein-coupled receptor (GPCR) superfamily, is at the core of the druggable proteome. In response to various endogenous agonists, such as photons, protons, ions, peptides, and proteins, GPCRs, which have about 800 members, bind and react. There is now a vast array of experimental structures of GPCRs, primarily in active form and with intracellular effectors, thanks to the cryo-EM resolution revolution. However, it is still difficult and rare to find high-resolution structures with tiny compounds, particularly antagonists and inverse agonists. To evaluate the state of the art of computational modeling and docking for the GPCR superfamily, the GPCR Dock competitions were created.
The Purpose and Impact of GPCR Dock Challenges
GPCR Dock challenges the community to make blind predictions of complex geometries from amino acid sequences by leveraging recently resolved and as-yet-unpublished experimental structures of receptor-ligand complexes. One important measure of the progress of protein modeling in the druggable proteome is the results of GPCR Dock evaluations.
With its emphasis on SMO receptors and other proteins, the GPCR Dock 2021 competition reflects a dynamic evolution in GPCR research. Strong target-template sequence alignments, accurate loop geometry predictions, and a variety of binding pocket identifications are the issues that the competition seeks to solve. A deep learning algorithm for protein structure prediction called AlphaFold2 has surfaced as a promising solution to these problems. Despite its promise, AlphaFold2 has GPCR-specific drawbacks. The competition seeks to overcome AlphaFold’s constraints and difficulties by utilizing these developments, experimental discoveries, and computational breakthroughs to shed light on a revolutionary route for GPCR structural modeling and docking.
Increased Complexity and Target Diversity in GPCR Dock 2021
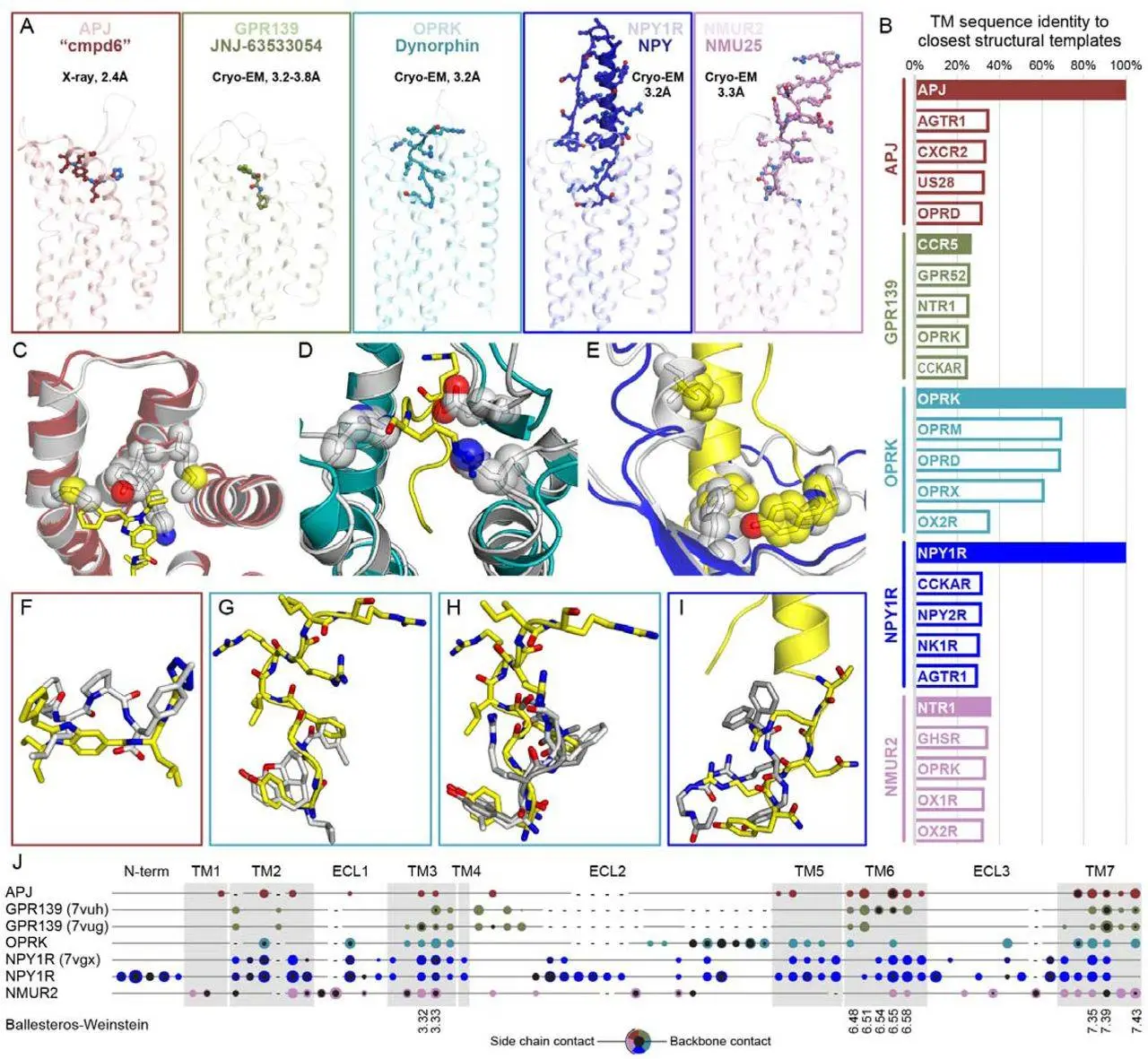
Compared to earlier evaluations, the 4th GPCR Dock assessment in 2021 included a greater number of targets, including 5 target complexes of 5 receptors. The evaluation did not include peptide complexes, which are typically difficult prediction targets. While the other two receptors had low-level homology structures, three GPCR Dock 2021 target receptors have experimental structures in the PDB. GPCRs’ orthosteric binding sites show a great deal of conformational variability, underscoring the difficulties in molecular recognition within the GPCR family. If the target complex is sufficiently conformationally different, homologous structures can bias modeling attempts even though they may offer essential modeling templates. GPCRs without preexisting structures posed a unique problem that necessitated the development and improvement of creative modeling strategies. Because more computational methods without preset structural templates can be explored, this could result in more accurate predictions.
In contrast to previous contests, the state of the art was significantly improved in GPCR Dock 2021 to offset an overall increase in the difficulty of prediction problems. The PDB now contains a considerably greater general grasp of GPCR conformation and ligand interaction principles because of the many more GPCR structures from different families that have been solved in both active and inactive states. With little assistance from human experts, AlphaFold2, which was released just a few months prior to the evaluation, quickly established itself as the standard method for incorporating these ideas into the models. This led to a significant improvement in the ability to accurately anticipate receptor architectures and complexes with bigger peptides. Induced fit was still a major problem for these complexes, though, and difficulties with shorter peptides and tiny molecules persisted.
Impact of AlphaFold2 and Challenges in Small-Molecule Docking
The success of GPCR complex predictions, both with and without near homologs in the PDB, was found to be significantly influenced by AlphaFold2, according to the GPCR Dock 2021 study. There is a weak correlation between the training set’s homologous receptor representation and the precision of AF2 predictions for GPCRs. With the OPRK/Dynorphin complex being a difficult target because of its lack of co-evolution, co-folding techniques significantly aided predictions for peptide complexes. The drawbacks of AF2 for small-molecule complexes that needed compound docking included reduced prediction accuracy for receptor loops and flexible sections, as well as the inability to predict receptor conformational ensembles and separate functional states. These findings are consistent with earlier research that compared AF2 to template-based techniques or assessed how well AF2 models replicate small-molecule compounds in virtual screening or docking.
Conclusion
Deep learning models to predict protein structures with nonpeptidic ligands, such as small-molecule compounds, have been developed by academia and businesses since 2021. In this endeavor, the 2024 release of AlphaFold3 (AF3) and further diffusion-based models for protein co-folding with tiny ligands has proved crucial. These models can still be limited in their ability to forecast complexes that lack chemistry or homology to the structures in the training set. Even in the age of AlphaFold2/3, expert knowledge, complex SAR data, and traditional model improvement remain essential for precise predictions. Both structure-based drug discovery and traditional computational modeling and refinement still require experimental high-resolution GPCR structure determination, particularly with small-molecule chemicals, even though computationally generated models of GPCR complexes with peptides can be more accurate than low-resolution experimental structures.
Article Source: Reference Paper
Disclaimer:
The research discussed in this article was conducted and published by the authors of the referenced paper. CBIRT has no involvement in the research itself. This article is intended solely to raise awareness about recent developments and does not claim authorship or endorsement of the research.
Important Note: bioRxiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Follow Us!
Learn More:




Deotima is a consulting scientific content writing intern at CBIRT. Currently she's pursuing Master's in Bioinformatics at Maulana Abul Kalam Azad University of Technology. As an emerging scientific writer, she is eager to apply her expertise in making intricate scientific concepts comprehensible to individuals from diverse backgrounds. Deotima harbors a particular passion for Structural Bioinformatics and Molecular Dynamics.











{kind=link}