
The process of drug discovery has always necessitated a large amount of time and effort as it requires a careful combination of both chemical and biological expertise. Lately, however, the introduction of artificial intelligence and machine learning has changed this paradigm. One such advancement is Rag2Mol, a novel model that aims to improve and facilitate structure-based drug design (SBDD). This was undertaken by a team of researchers led by Peidong Zhang and collaborators. This study marks a tremendous breakthrough in the evolution of computational drug design. The team, which included members from Chinese and other research centers, created Rag2Mol in response to the urgent problem of a rapid, accurate, and economical drug discovery process. Their motivations have been the challenges of the current state of the art and the need for a more versatile model that applies biochemical knowledge and supercomputing.
Understanding the Foundations of Rag2Mol
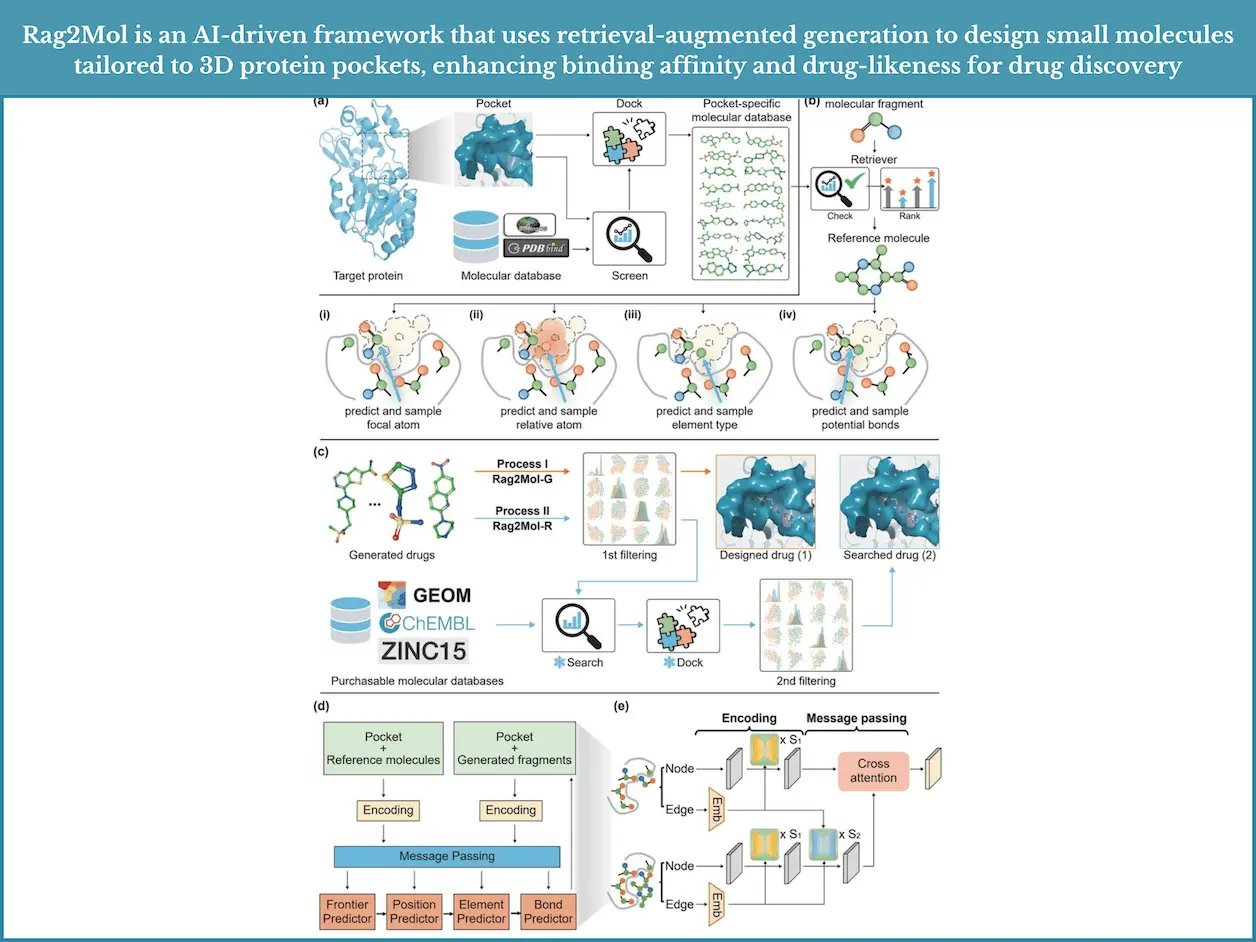
The principle of retrieval-augmented generation (RAG), which is at the core of this innovation, is also what Rag2Mol employs to transform the process of drug design. The rationale behind RAG, however, is to develop machine learning models whose performance can be augmented through the use of supplemented information retrieved during the training and inference periods. The model ‘internalizes’ messages from some databases and uses this when predicting new drug candidates.
In this context of drug design and particular structure-based drug design (SBDD), the objective is to develop molecules that can bind effectively to select cavities of specific proteins. One of the major reasons for this challenge is the geometrical and biological complexities related to this task encountered with conventional approaches. Rag2Mol addresses these issues by integrating acquired knowledge from biochemical databases into the process of generating novel 3D molecular shapes. This positions Rag2Mol in such a way that it can design molecules that meet the requirements of not only fitting into the protein pocket but also having high affinity, low toxicity, and ease of synthesis.
The Problem with Traditional Drug Design Approaches
It is a well-known fact that drug discovery is hard. Especially novel, small-molecule drug discoverers usually have a mentor’s taste or a good financial plan. Here, a scientist’s imagination is well needed: finding a molecule that has a decent binding interaction with the target is only a primitive stage. Apart from that, a molecule needs to have other important properties: it should be easy to synthesize, be reasonably stable, and register minimum side effects. Sometimes, many SBDD models are based on a plethora of chemical and biological interaction databases, but, more often than not, the three-dimensional geometric and natural biochemical ones of the target proteins of interest are completely overlooked.
- SDBD systems, as most other models currently applied in drug discovery, have two principal disadvantages:
- Scope Limitations: They polarize or tone down to the specific data they were exposed to during training and, therefore, cannot extend their application to new chemical spaces or create new structural molecular models that can meet all the functional requirements of the desired object.
- Geometric Constraints: It is a well-known fact that these molecular interactions have a three-dimensional character, which is a hard thing to show, while most SBDD techniques fail to reliably position a molecular structure in the target protein pocket, and so yield unsuitable drug candidates.
Rag2Mol was designed specifically to address the earlier restrictions by implementing retrieval-based learning, which makes it possible to gather more biochemically relevant information while still preserving geometric fidelity.
How Rag2Mol Works
Rag2Mol adopts a retrieval-augmented generation paradigm for the design of new molecules. There’s also a crucial step where the model employs structural templates and known interactions as guiding information for new candidates. This kind of input is stored outside of the model, for example, in databases, and can be accessed while the model learns. The more the model retrieves during design, the more accurate and appropriate drug candidates it can create. Designing new drugs. Progress is made from identifying functions to identifying drugs.
Training: Trains the model using graphs. From the known interaction graph, the model one-shot learns the interaction, which allows the model to learn functional components within a larger interaction landscape.
Generation: Rag2Mol formulates the drug, creating an understanding of how the molecules should interact with biological targets. It is very important that the final construction of the molecule happens by other molecules that provide fundamental interactions. Extending the understanding of activity-predicting how likely the molecule will exert the desired effects on living systems.
The biggest challenge is bridging the gap between comprehension, identification, and engineering. In general, imposing narrowing design principles represents a shift from creation to identification. More so, it’s easier to say than done. Featuring state-of-the-art provisions and generally outperforms current rough methods.
Experimental Results and Performance
The researchers employed Rag2Mol in different SBDD and virtual screening tasks as well and assessed its results against some existing solutions such as Pocket2Mol, GraphBP, FLAG, and TargetDiff. These models were also cross-validated in the CrossDock 2020 dataset, which is a standard in the field of drug development.
Metrics Used: A set of metrics that are largely established in the scientific community were employed by the researchers to evaluate Rag2Mol’s performance as follows
Vina Dock and Vina Score: They evaluate the binding energy of molecules pre- and post-docking simulations.
QED (Quantitative Estimation of Drug-likeness): It is an index that quantifies the degree to which the molecules that are created conform to the characteristics of drugs.
SA (Synthetic Accessibility): This indicates the ease with which a particular molecule can be synthesized in the lab.
Lipinski’s Rule of Five: This explains the potential of a molecule to be ingested and act as a drug in a human being.
RMSD (Root Mean Square Deviation): It provides a measure of the precision of molecular poses that are generated by the model.
Results: On the other hand, Rag2Mol showed better results in almost all metrics whenever it was compared with existing models. Out of those, Notably, over 60% of molecules generated by Rag2Mol exhibited stronger binding energies than the test ligands, a result that highlights the model’s exceptional accuracy. Besides, Rag2Mol was able to generate molecules with more diversity, and hence, a wider range of drug candidates was obtained. With regards to synthetic accessibility and drug-like characteristics, Rag2Mol generated molecules that were more likely to be viable drug candidates than those produced by competing models.
Case Study: Tackling the “Undruggable” Protein
The study incorporated Rag2Mol into a difficult real-world application: designing molecules aimed at PTPN2, a protein deemed “undruggable” for the longest time because of its complicated architecture. Rag2Mol was able to produce two drug candidates, G1 and R1, which exhibited higher binding affinities than those of AC484, a known inhibitor. Not only did these molecules bind within the target protein pocket, but they were also more stable and easier to synthesize than other drugs.
Conclusion: A New Age for Drug Discovery
The Model Rag2Mol is a development worth noting in drug discovery. With retrieval-augmented generation, the method is efficient and scalable in terms of structure-based drug design. The model’s ability to bulk up with specific biochemical information at both training and generation processes gives it advantages over orthodox methods, allowing it to create a better variety of candidates that are more potent and easier to synthesize.
The successful cases of real-world trials, such as the Rag2Mol application in PTPN2, show its promise in addressing even the more difficult challenges in drug discovery. New cases for the development of AI-powered models such as Rag2Mol can bring more than just new software; they can also bring revolutionary ideas in the search for new therapies for diseases that have long eluded effective treatment.
Article Source: Reference Paper | Rag2Mol code is available on GitHub.
Important Note: bioRxiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Follow Us!
Learn More:




Anchal is a consulting scientific writing intern at CBIRT with a passion for bioinformatics and its miracles. She is pursuing an MTech in Bioinformatics from Delhi Technological University, Delhi. Through engaging prose, she invites readers to explore the captivating world of bioinformatics, showcasing its groundbreaking contributions to understanding the mysteries of life. Besides science, she enjoys reading and painting.











{kind=link}