
The subject of protein engineering has shown great promise for protein language models. Unfortunately, the main size at which the existing protein language models function is the residue, which restricts the information that they can convey at the atom level. Because of this restriction, we are unable to fully utilize the potential of protein language models for applications involving tiny chemicals and proteins. This work presents ESM-AA (ESM All-Atom), a unique method that allows for unified molecular modeling at both the atom and residue scales. Through the use of molecular modeling, ESM-AA outperforms existing techniques in tasks involving protein molecules by pretraining on multi-scale code-switch protein sequences and assembling residue-atom correlations. Protein language models are fully utilized by this unified molecular modeling, which exhibits both molecular knowledge acquisition and retention of its understanding of proteins.
Introduction
The application of Protein Language Models (PLMs), which allow the prediction of protein structure and fitness, has greatly advanced protein engineering. Protein design, fitness prediction, and structural prediction have made great progress thanks in large part to PLMs like ESM. Protein engineering has been transformed by these models, which include LM-Design and ESM-Fold. PLMs have limited potential for applications involving macromolecules and small molecules because they lack information at the atom scale, which is largely relevant to protein residue analysis. This restriction hampers their complete use in downstream applications. External small molecule models need to be incorporated in order to handle this. But modelling proteins only at the residue size would lead to low resolution, which would restrict the range of possible uses. Consequently, PLMs’ range of applications would be increased by extending them to the residue and atom scales.
Challenges Faced in the Development of Multi-scale PLMs
- The first difficulty lies in the incompatible vocabularies employed at the residue and atom scales, which make it difficult to achieve unified molecular modeling that functions well at both of these scales. Using atom-scale representation and pre-training of proteins rather than the original residue-scale pre-training is one possible way to introduce atomic information into PLMs. To be clear, a normal protein can have thousands of residues, each having hundreds of thousands of atoms. As a result, modeling using this approach would be ineffective.
- Second, it is not easy to develop a suitable position encoding that precisely describes the interactions between residues and atoms within the same protein. These relationships can change between residues and atoms.
Understanding ESM-AA (ESM All-Atom)
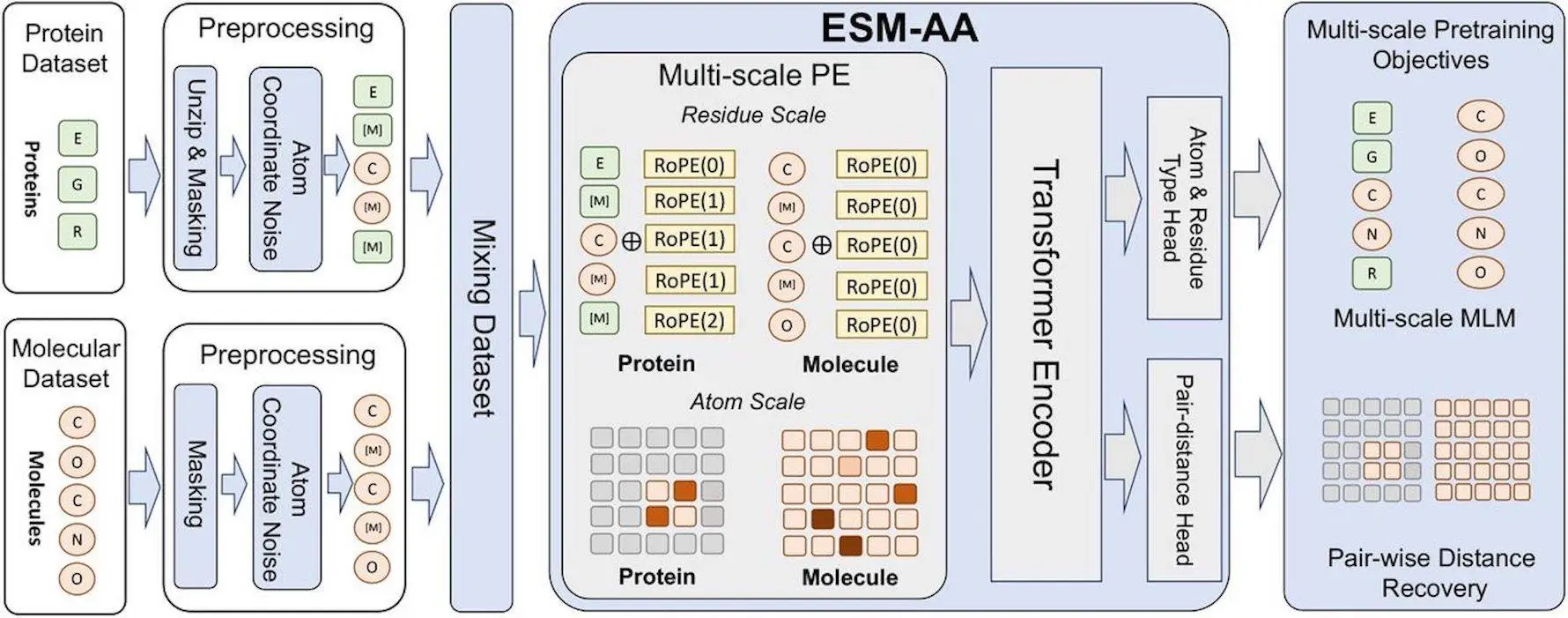
In order to address the issues mentioned above, the authors of this paper propose ESM-AA (ESM All-Atom), which enables multiscale unified molecular modeling by (i) pre-training on multi-scale code-switch protein sequences and (ii) using a multi-scale position encoding to describe the relationships between residues and atoms. Protein sequences are combined with multilingual code-switching in the notion of the machine translation tool ESMAA to learn multi-scale knowledge. Protein residues are randomly unzipped into atoms, and coordinates are assigned to create these sequences, which are a hybrid of sequence and structural data. ESMAA performs well with residue sequences and atomic coordinates, which improves machine translation.
To distinguish between atoms and residues in the code-switch sequence, ESM-AA employs a multi-scale position encoding. The original position encoding adheres to modern best practices at the residue scale, preventing unclear information from being conveyed across scales. All relationships within the code-switch sequence are efficiently described at the atom scale by a spatial distance matrix that directly represents 3D coordinates among unzipped atoms. In protein-molecule tasks, the unified molecular modeling of ESM-AA, a task model, performs better than earlier techniques. With the help of a wide range of benchmarks, the model learns molecular information without sacrificing its comprehension of proteins. When compared to established molecular benchmarks, the unified molecular modeling also performs better than various molecule-specific models, demonstrating the promise of this technique in protein-molecule applications.
ESM-AA Retains Strong Ability of Understanding Proteins
The potential of a protein pre-training model called ESM-AA to retain a thorough knowledge of proteins is being evaluated. Tasks that need models to understand local protein structures like helices and strands, like secondary structure prediction and contact prediction, are used to test the model. For these tasks, the input for the baseline and ESM-AA approaches is the same. The outcomes demonstrate that ESM-AA does not lose its grasp of proteins since it outperforms ESM-2 in both secondary structure prediction and contact prediction. The work demonstrates the potential of ESM-AA for enhanced protein comprehension by indicating that it can do so by initializing its settings with a larger ESM-2.
Conclusion
Researchers provide a multi-scale protein language model in this work called ESM-AA. This model uses a multi-scale position encoding to describe interactions between residues and atoms and pre-trains on multi-scale code-switch protein sequences to realize multi-scale unified molecular modeling. The outcomes of the experiment demonstrate that ESM-AA performs better than earlier techniques in protein-molecule tasks and successfully incorporates molecular knowledge into the protein language model without compromising the comprehension of proteins.
Article Source: Reference Paper
Important Note: bioRxiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Follow Us!
Learn More:




Deotima is a consulting scientific content writing intern at CBIRT. Currently she's pursuing Master's in Bioinformatics at Maulana Abul Kalam Azad University of Technology. As an emerging scientific writer, she is eager to apply her expertise in making intricate scientific concepts comprehensible to individuals from diverse backgrounds. Deotima harbors a particular passion for Structural Bioinformatics and Molecular Dynamics.











{kind=link}