
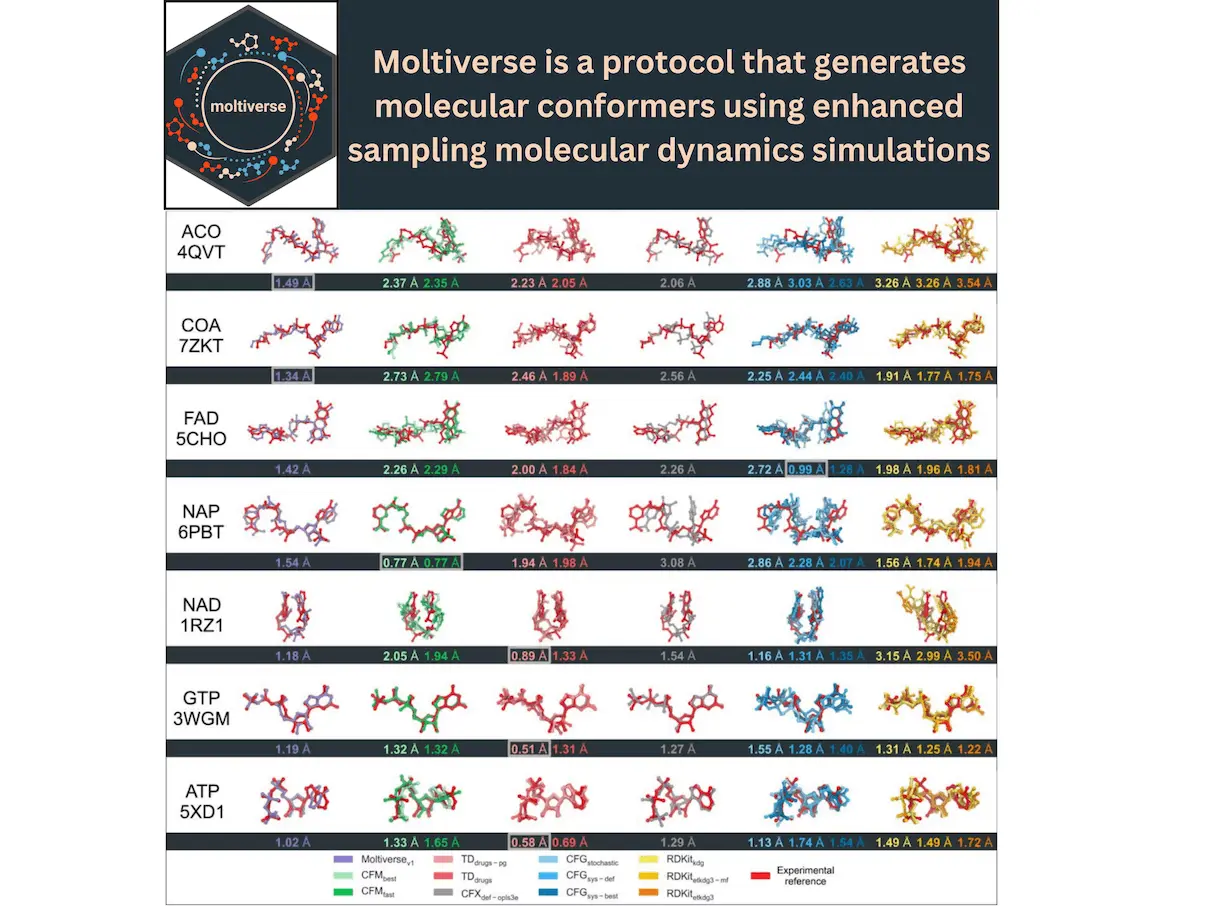
For drug development to be effective, it is essential to accurately predict the various bound-state conformations of small compounds, especially in situations where intricate protein-ligand interactions are unknown. Although there are established techniques, it is still difficult to efficiently explore the large conformational space. In this work, researchers present Moltiverse, a novel methodology for conformer generation that uses enhanced sampling models of molecular dynamics (MD). The extended adaptive biasing force (eABF) algorithm and metadynamics led by a single collective variable (RDGYR) are used by Moltiverse, a conformer generation algorithm, to effectively traverse the conformational landscape of molecules. Moltiverse outperforms well-known programs like Torsional Diffusion, Conformator, RDKit, CONFORGE, and ConfGenX in terms of accuracy and competitiveness. There are hundreds to thousands of experimental conformers per molecule in the Cofactorv1 dataset, a supplementary resource for evaluating conformer generators, but only seven small molecule cofactors. This presents a problem for benchmarks for conformer production. When it comes to conformer generation techniques like Moltiverse, this dataset pushes the limits of accuracy and diversity.
Introduction
Drug discovery techniques, including virtual screening, structure-based drug design, molecular docking, and pharmacophore modeling, all depend on the precise prediction of small molecule bound-like conformations. Efficient exploration of the extensive conformational space of flexible molecules is a problem, particularly when the bioactive configuration is unknown or deviates greatly from low-energy states. Both open-source and commercial conformator generation tools provide a wide range of approaches that strike a compromise between geometry correctness, accuracy, precision, diversity, and computing efficiency. Balloon, Confab, Conforge, FROG2, OpenBabel, and RDKit are traditional open-source conformer generators that provide transparency and flexibility in their processes. High-performance algorithms are available through commercial programs such as CAESAR, ConfGen, Conformator, COSMOS, ForceGen, and OMEGA.
With the use of sophisticated machine learning techniques, artificial intelligence models for conformer generation have recently surfaced, predicting molecular conformations. With the potential to transform the field, these AI-driven techniques promise to bring together the precision of physics-based methods with the speed of knowledge-based approaches. With an emphasis on molecules with a reasonable number of rotatable bonds, the RDKit system employs experimental data to investigate the torsional space of molecules.
For molecules with a moderate number of rotatable bonds, this approach works well. It is better to use stochastic sampling approaches for molecules that are very flexible. Higher and lower distance bounds between atom pairs are defined by the distance geometry (DG) technique in RDKit, which is based on structural constraints and chemical information. By choosing distances within these constraints at random for every pair of atoms, conformations are created. 3D coordinates are then produced via embedding. Libraries of pre-calculated fragment conformations and torsion angles are included in knowledge-based methods.
Understanding Moltiverse
To investigate the conformational landscape of small molecules, researchers present Moltiverse, a novel approach for conformer creation that uses enhanced sampling molecular dynamics (MD) simulations. Fundamentally, Moltiverse integrates metadynamics and the extended adaptive biasing force (eABF) method, using the radius of gyration (RDGYR) as a single collective variable. Energy barriers that frequently confine conventional MD simulations in local minima are overcome by this special combination, which allows efficient conformer ensembles in a short simulation time. A physics-based exploration of conformational space is made possible by Moltiverse’s MD-based method, unlike stochastic approaches that depend on random sampling or systematic search methods that thoroughly investigate torsional space. Traditional approaches may find it difficult to efficiently sample pertinent conformations in molecules with complex energy landscapes or many rotatable bonds. In these above cases, this approach is especially beneficial.
Conformer generation protocol of Moltiverse
Moltiverse is developed in the contemporary Crystal language and is a ligand conformer generator. With the chem.cr library serving as its basis for file conversion, input creation, calculation execution, geometric analysis, and clustering, Moltiverse leverages the strong ecosystem of open-source programs to process the molecules and carry out the conformational sampling. There are seven major steps in the conformer-generating protocol: molecular pre-processing, which includes using Open Babel software to convert the SMILES code into three-dimensional coordinates; (ii) initial conformer extension; (iii) parameterizing the molecule using the GAFF2 force field with AmberTools, (iv) energetic minimization, (v) sampling the molecule using the M-eABF method in vacuum using the NAMD molecular dynamics engine, (vi) structure clustering, and (vii) refining of the conformer ensemble by electronic structure optimization simulations using XTB software.
Conclusion
Moltiverse is a potent tool for producing high-quality, bound-like structures across various compounds. It is built on open-source apps and the M-eABF technique. It is especially useful for larger, more flexible molecules since it employs the radius of gyration as a collective variable for conformal sampling. Regarding RMSDhigh and RMSDrange metrics, Moltiverse performs similarly to Conformator and, in tandem with Conformge, achieves the best RMSD-RDGYRcorr values. It is a useful technique in computational chemistry and drug development because it produces conformers with low energy and few processing errors, particularly for complicated, flexible compounds.
Article Source: Reference Paper | The Moltiverse source code is available on GitHub.
Disclaimer:
The research discussed in this article was conducted and published by the authors of the referenced paper. CBIRT has no involvement in the research itself. This article is intended solely to raise awareness about recent developments and does not claim authorship or endorsement of the research.
Important Note: ChemRxiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Follow Us!
Learn More:




Deotima is a consulting scientific content writing intern at CBIRT. Currently she's pursuing Master's in Bioinformatics at Maulana Abul Kalam Azad University of Technology. As an emerging scientific writer, she is eager to apply her expertise in making intricate scientific concepts comprehensible to individuals from diverse backgrounds. Deotima harbors a particular passion for Structural Bioinformatics and Molecular Dynamics.











{kind=link}